Introduction to Nextflow
Introduction to Nextflow
DSL for data-driven computational pipelines. www.nextflow.io.
What is Nextflow?
Nextflow is a domain specific language (DSL) for workflow orchestration that stems from Groovy. It enables scalable and reproducible workflows using software containers. It was developed at the CRG in the Lab of Cedric Notredame by Paolo Di Tommaso. The Nextflow documentation is available here and you can ask help to the community using their gitter channel
In 2020, Nextflow has been upgraded from DSL1 version to DSL2. In this course we will use exclusively DSL2.
What is Nextflow for?
It is for making pipelines without caring about parallelization, dependencies, intermediate file names, data structures, handling exceptions, resuming executions, etc.
It was published in Nature Biotechnology in 2017.

There is a growing number of PubMed publications citing Nextflow.

A curated list of Nextflow pipelines.
Many pipelines written collaboratively are provided by the NF-core project.
Some pipelines written in Nextflow have been used for the SARS-Cov-2 analysis, for example:
The artic Network pipeline ncov2019-artic-nf.
The CRG / EGA viral Beacon pipeline Master of Pores.
The nf-core pipeline viralrecon.
Main advantages
Fast prototyping
You can quickly write a small pipeline that can be expanded incrementally. Each task is independent and can be easily added to other. You can reuse scripts without re-writing or adapting them.
Reproducibility
Nextflow supports Docker and Singularity containers technology. Their use will make the pipelines reproducible in any Unix environment. Nextflow is integrated with GitHub code sharing platform, so you can call directly a specific version of a pipeline from a repository, download and use it on-the-fly.
Portability
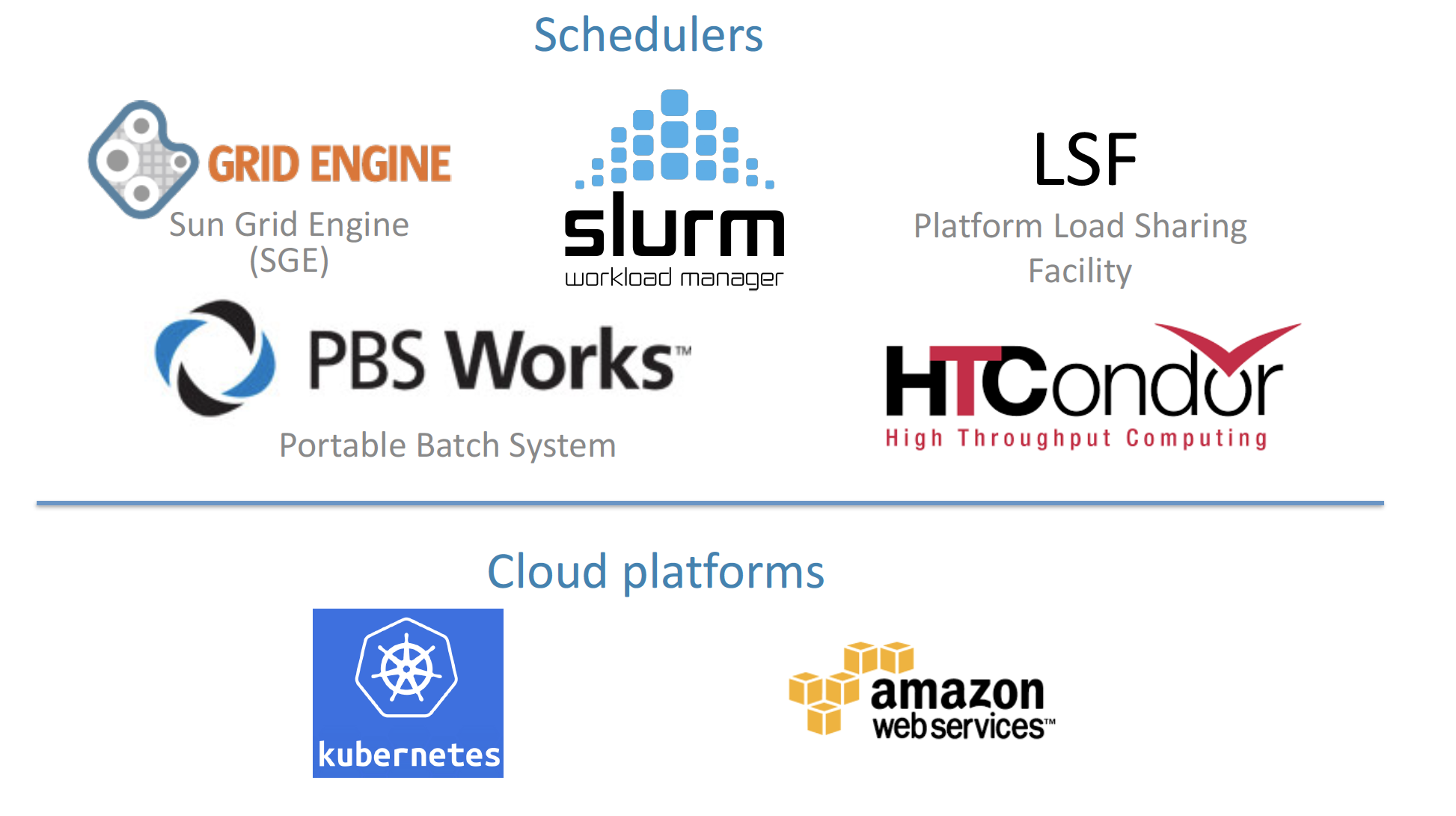
Nextflow can be executed on multiple platforms without modifiying the code. It supports several schedulers such as SGE, LSF, SLURM, PBS, HTCondor and cloud platforms like Kubernetes, Amazon AWS, Google Cloud.
Scalability
Nextflow is based on the dataflow programming model which simplifies writing complex pipelines. The tool takes care of parallelizing the processes without additionally written code. The resulting applications are inherently parallel and can scale-up or scale-out transparently; there is no need to adapt them to a specific platform architecture.
Resumable, thanks to continuous checkpoints
All the intermediate results produced during the pipeline execution are automatically tracked. For each process a temporary folder is created and is cached (or not) once resuming an execution.
Workflow structure
The workflows can be represented as graphs where the nodes are the processes and the edges are the channels. The processes are blocks of code that can be executed - such as scripts or programs - while the channels are asynchronous queues able to connect processes among them via input / output.
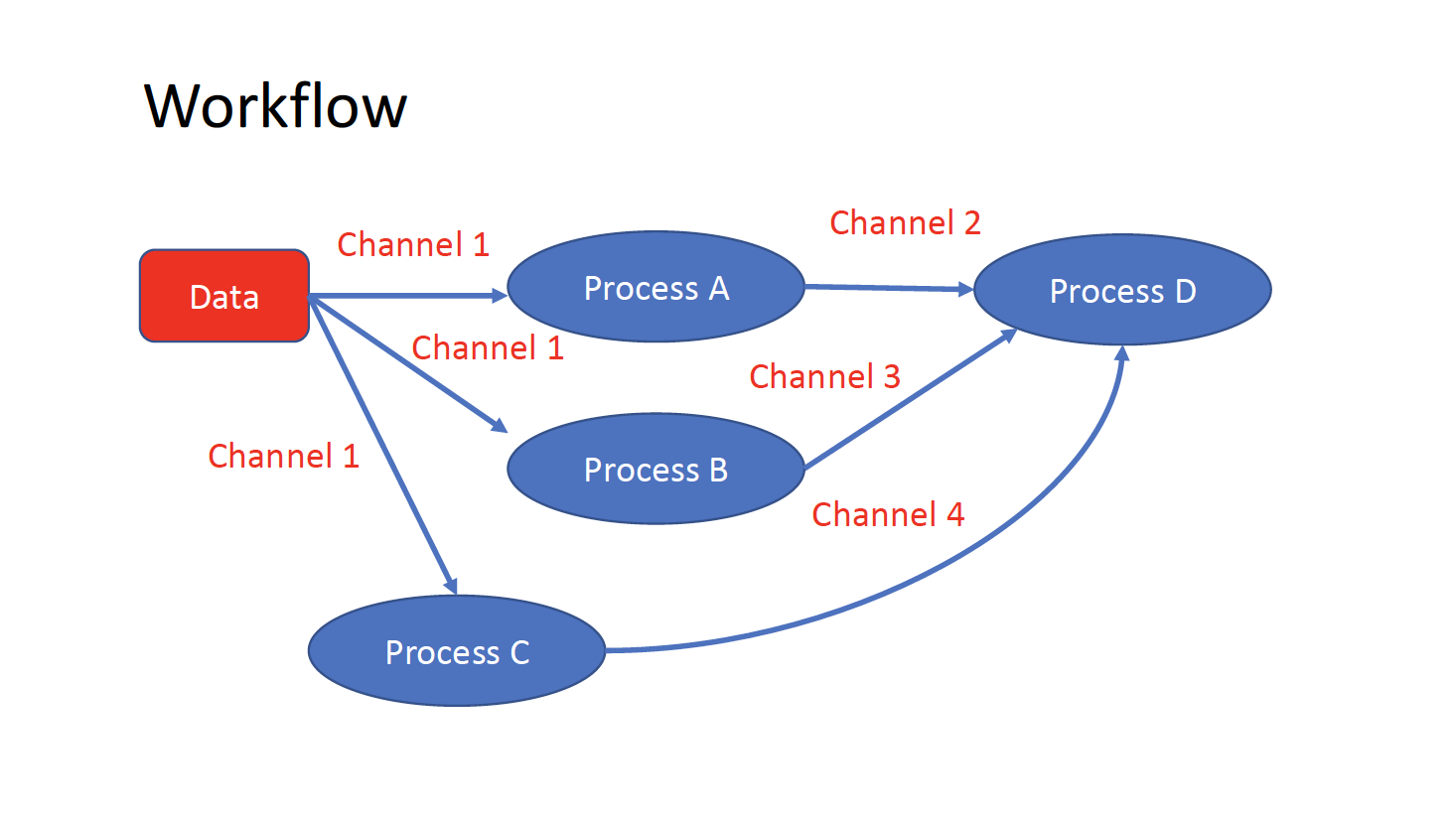
Processes are independent from each another and can be run in parallel, depending on the number of elements in a channel. In the previous example, processes A, B and C can be run in parallel and only when they ALL end the process D is triggered.
Installation
Note
Nextflow is already installed on the machines provided for this course. You need at least the Java version 8 for the Nextflow installation.
Tip
You can check the version fo java by typing:
java -version
Then we can install Nextflow with:
curl -s https://get.nextflow.io | bash
This will create the nextflow executable that can be moved, for example, to /usr/local/bin.
We can test that the installation was successful with:
nextflow run hello
N E X T F L O W ~ version 20.07.1
Pulling nextflow-io/hello ...
downloaded from https://github.com/nextflow-io/hello.git
Launching `nextflow-io/hello` [peaceful_brahmagupta] - revision: 96eb04d6a4 [master]
executor > local (4)
[d7/d053b5] process > sayHello (4) [100%] 4 of 4 ✔
Ciao world!
Bonjour world!
Hello world!
Hola world!
This command downloads and runs the pipeline hello.
We can now launch a test pipeline:
nextflow run nextflow-io/rnaseq-nf -with-singularity
This command will automatically pull the pipeline and the required test data from the github repository
The command -with-singularity will automatically trigger the download of the image nextflow/rnatoy:1.3 from DockerHub and convert it on-the-fly into a singularity image that will be used for running each step of the pipeline.
The pipeline can also recognize the queue system which is used on the machine where it is launched. In the following examples, I launched the same pipeline both on the CRG high performance computing (HPC) cluster and on my MacBook:
The result from CRG HPC:
nextflow run nextflow-io/rnaseq-nf -with-singularity
N E X T F L O W ~ version 21.04.3
Pulling nextflow-io/rnaseq-nf ...
downloaded from https://github.com/nextflow-io/rnaseq-nf.git
Launching `nextflow-io/rnaseq-nf` [serene_wing] - revision: 83bdb3199b [master]
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: /users/bi/lcozzuto/.nextflow/assets/nextflow-io/rnaseq-nf/data/ggal/ggal_1_48850000_49020000.Ggal71.500bpflank.fa
reads : /users/bi/lcozzuto/.nextflow/assets/nextflow-io/rnaseq-nf/data/ggal/*_{1,2}.fq
outdir : results
[- ] process > RNASEQ:INDEX -
[- ] process > RNASEQ:FASTQC -
executor > crg (6)
[cc/dd76f0] process > RNASEQ:INDEX (ggal_1_48850000_49020000) [100%] 1 of 1 ✔
[7d/7a96f2] process > RNASEQ:FASTQC (FASTQC on ggal_liver) [100%] 2 of 2 ✔
[ab/ac8558] process > RNASEQ:QUANT (ggal_gut) [100%] 2 of 2 ✔
[a0/452d3f] process > MULTIQC [100%] 1 of 1 ✔
Pulling Singularity image docker://quay.io/nextflow/rnaseq-nf:v1.0 [cache /nfs/users2/bi/lcozzuto/aaa/work/singularity/quay.io-nextflow-rnaseq-nf-v1.0.img]
WARN: Singularity cache directory has not been defined -- Remote image will be stored in the path: /nfs/users2/bi/lcozzuto/aaa/work/singularity -- Use env variable NXF_SINGULARITY_CACHEDIR to specify a different location
Done! Open the following report in your browser --> results/multiqc_report.html
Completed at: 01-Oct-2021 12:01:50
Duration : 3m 57s
CPU hours : (a few seconds)
Succeeded : 6
The result from my MacBook:
nextflow run nextflow-io/rnaseq-nf -with-docker
N E X T F L O W ~ version 21.04.3
Launching `nextflow-io/rnaseq-nf` [happy_torvalds] - revision: 83bdb3199b [master]
R N A S E Q - N F P I P E L I N E
===================================
transcriptome: /Users/lcozzuto/.nextflow/assets/nextflow-io/rnaseq-nf/data/ggal/ggal_1_48850000_49020000.Ggal71.500bpflank.fa
reads : /Users/lcozzuto/.nextflow/assets/nextflow-io/rnaseq-nf/data/ggal/*_{1,2}.fq
outdir : results
executor > local (6)
[37/933971] process > RNASEQ:INDEX (ggal_1_48850000_49020000) [100%] 1 of 1 ✔
[fe/b06693] process > RNASEQ:FASTQC (FASTQC on ggal_gut) [100%] 2 of 2 ✔
[73/84b898] process > RNASEQ:QUANT (ggal_gut) [100%] 2 of 2 ✔
[f2/917905] process > MULTIQC [100%] 1 of 1 ✔
Done! Open the following report in your browser --> results/multiqc_report.html
Nextflow main concepts.
Channels and Operators
There are two different types of channels:
A queue channel is a non-blocking unidirectional FIFO (First In, First Out) queue which connects two processes or operators.
A value channel, a.k.a. singleton channel, is bound to a single value and can be read unlimited times without consuming its content.
An operator is a method that reshapes or connects different channels applying specific rules.
We can write a very simple Nextflow script: save the following piece of code in the test0.nf file:
#!/usr/bin/env nextflow
// This is a comment
/*
* This is a block of comments
*/
// This is needed for activating the new DLS2
nextflow.enable.dsl=2
//Let's create a channel from string values
str = Channel.from('hello', 'hola', 'bonjour')
/*
* Let's print that channel using the operator view()
* https://www.nextflow.io/docs/latest/operator.html#view
*/
str.view()
Once the file is saved, execute it with:
nextflow run test0.nf
N E X T F L O W ~ version 20.07.1
Launching `test0.nf` [agitated_avogadro] - revision: 61a595c5bf
hello
hola
bonjour
As you can see, the Channel is just a collection of values, but it can also be a collection of file paths.
Let’s create three empty files with the touch command:
touch aa.txt bb.txt cc.txt
and another script (test2.nf) with the following code:
#!/usr/bin/env nextflow
// enable DSL2
nextflow.enable.dsl=2
/*
* Let's create the channel `my_files`
* using the method fromPath
*/
Channel
.fromPath( "*.txt" )
.set {my_files}
// We can use the view() operator again to see the content of channel "my_files"
my_files.view()
We can now execute test2.nf:
nextflow run test2.nf
N E X T F L O W ~ version 20.07.1
Launching `test2.nf` [condescending_hugle] - revision: f513c0fac3
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/aa.txt
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/bb.txt
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/cc.txt
Once executed, we can see that a folder named work is generated. Nextflow stores in this folder the intermediate files generated by the processes.
EXERCISE
Create a couple of files (e.g., paired-end reads) and read them as a tuple.
First, create a couple of empty files:
touch aaa_1.txt aaa_2.txt
See here fromFilePairs.
Solution
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
/*
* Let's create the channel `my_files`
* using the method fromFilePairs
*/
Channel
.fromFilePairs( "aaa_{1,2}.txt" )
.set {my_files}
my_files.view()
For the second part of this exercise, We can start again from .fromPath and read the previous 3 .txt files (“aa.txt”, “bb.txt”, “cc.txt”) into the input channel.
- Reshape the input channel using different operators by generating:
A single emission.
A channel with each possible file combination
A tuple with a custom id, i.e. something like [“id”, [“aa.txt”, “bb.txt”, “cc.txt”]]
See here the list of Operators available.
Solution
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
Channel
.fromPath("{aa,bb,cc}.txt")
.set {my_files}
my_files
.collect()
.view()
// You can also write it as: my_files.collect().view()
my_files
.combine(my_files)
.view()
my_files
.collect()
.map{
["id", it]
}
.view()
Processes
Let’s add a process to the previous script test0.nf and let’s call it test1.nf
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
str = Channel.from('hello', 'hola', 'bonjour')
/*
* Creates a process which receives an input channel containing values
* Each value emitted by the channel triggers the execution
* of the process. The process stdout is captured and sent over
* the another channel.
*/
process printHello {
tag { "${str_in}" } // this is for displaying the content of `str_in` in the log file
input:
val str_in
output:
stdout
script:
"""
echo ${str_in} in Italian is ciao
"""
}
The process can be seen as a function that is composed of:
An input part where the input channels are defined.
An output part where we specify what to store as a result, that will be sent to other processes or published as final result.
A script part where we have the block of code to be executed using data from the input channel, and that will produce the output for the ouput channel.
Any kind of code / command line can be run there, as it is language agnostic.
Note
You can have some trouble with escaping some characters: in that case, it is better to save the code into a file and call that file as a program.
Before the input, you can indicate a tag that will be reported in the log. This is quite useful for logging / debugging.
Workflow
The code above will produce nothing, because it requires the part that will actually call the process and connect it to the input channel.
This part is called a workflow.
Let’s add a workflow to our code:
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
str = Channel.from('hello', 'hola', 'bonjour')
process printHello {
tag "${str_in}"
input:
val str_in
output:
stdout
script:
"""
echo ${str_in} in Italian is ciao
"""
}
/*
* A workflow consists of a number of invocations of processes
* where they are fed with the expected input channels
* as if they were custom functions. You can only invoke a process once per workflow.
*/
workflow {
result = printHello(str)
result.view()
}
We can run the script sending the execution in the background (with the -bg option) and saving the log in the file log.txt.
nextflow run test1.nf -bg > log.txt
Nextflow log
Let’s inspect the log file:
cat log.txt
N E X T F L O W ~ version 20.07.1
Launching `test1.nf` [high_fermat] - revision: b129d66e57
[6a/2dfcaf] Submitted process > printHello (hola)
[24/a286da] Submitted process > printHello (hello)
[04/e733db] Submitted process > printHello (bonjour)
hola in Italian is ciao
hello in Italian is ciao
bonjour in Italian is ciao
The tag allows us to see that the process printHello was launched three times using the hola, hello and bonjour values contained in the input channel.
At the start of each row, there is an alphanumeric code:
**[6a/2dfcaf]** Submitted process > printHello (hola)
This code indicates the path in which the process is “isolated” and where the corresponding temporary files are kept in the work directory.
Note
IMPORTANT: Nextflow will randomly generate temporary folders so they will be named differently in your execution!!!
Let’s have a look inside that folder:
# Show the folder's full name
echo work/6a/2dfcaf*
work/6a/2dfcafc01350f475c60b2696047a87
# List was is inside the folder
ls -alht work/6a/2dfcaf*
total 40
-rw-r--r-- 1 lcozzuto staff 1B Oct 7 13:39 .exitcode
drwxr-xr-x 9 lcozzuto staff 288B Oct 7 13:39 .
-rw-r--r-- 1 lcozzuto staff 24B Oct 7 13:39 .command.log
-rw-r--r-- 1 lcozzuto staff 24B Oct 7 13:39 .command.out
-rw-r--r-- 1 lcozzuto staff 0B Oct 7 13:39 .command.err
-rw-r--r-- 1 lcozzuto staff 0B Oct 7 13:39 .command.begin
-rw-r--r-- 1 lcozzuto staff 45B Oct 7 13:39 .command.sh
-rw-r--r-- 1 lcozzuto staff 2.5K Oct 7 13:39 .command.run
drwxr-xr-x 3 lcozzuto staff 96B Oct 7 13:39 ..
You see a lot of “hidden” files:
.exitcode, contains 0 if everything is ok, another value if there was a problem.
.command.log, contains the log of the command execution. It is often identical to .command.out
.command.out, contains the standard output of the command execution
.command.err, contains the standard error of the command execution
.command.begin, contains what has to be executed before .command.sh
.command.sh, contains the block of code indicated in the process
.command.run, contains the code made by nextflow for the execution of .command.sh, and contains environmental variables, eventual invocations of linux containers etc.
For example, the content of .command.sh is:
cat work/6a/2dfcaf*/.command.sh
#!/bin/bash -ue
echo hola in Italian is ciao
And the content of .command.out is
cat work/6a/2dfcaf*/.command.out
hola in Italian is ciao
You can also name the workflows to combine them in the main workflow. For example, using this code you can execute two different workflows that contain the same process:
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
str = Channel.from('hello', 'hola', 'bonjour')
process printHello {
tag "${str_in}"
input:
val str_in
output:
stdout
script:
"""
echo ${str_in} in Italian is ciao
"""
}
/*
* A workflow can be named as a function and receive an input using the take keyword
*/
workflow first_pipeline {
take: str_input
main:
printHello(str_input).view()
}
/*
* You can re-use the previous processes and combine as you prefer
*/
workflow second_pipeline {
take: str_input
main:
printHello(str_input.collect()).view()
}
/*
* You can then invoke the different named workflows in this way
* passing the same input channel `str` to both
*/
workflow {
first_pipeline(str)
second_pipeline(str)
}
We can add the collect operator to the second workflow that would collect the output from different executions and return the resulting list as a sole emission.
Let’s run the code:
nextflow run test1.nf -bg > log2
cat log2
N E X T F L O W ~ version 20.07.1
Launching `test1.nf` [irreverent_davinci] - revision: 25a5511d1d
[de/105b97] Submitted process > first_pipeline:printHello (hello)
[ba/051c23] Submitted process > first_pipeline:printHello (bonjour)
[1f/9b41b2] Submitted process > second_pipeline:printHello (hello)
[8d/270d93] Submitted process > first_pipeline:printHello (hola)
[18/7b84c3] Submitted process > second_pipeline:printHello (hola)
hello in Italian is ciao
bonjour in Italian is ciao
[0f/f78baf] Submitted process > second_pipeline:printHello (bonjour)
hola in Italian is ciao
['hello in Italian is ciao\n', 'hola in Italian is ciao\n', 'bonjour in Italian is ciao\n']
EXERCISE
Change the pipeline to produce files instead of standard output.
You can write another process to handle a list in the workflow2 (workflow second_pipeline). You also need to specify in the workflow the output using the **emit** keyword.
Solution
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
str = Channel.from('hello', 'hola', 'bonjour')
process printHello {
tag "${str_in}"
input:
val str_in
output:
path("${str_in}.txt")
script:
"""
echo ${str_in} in Italian is ciao > ${str_in}.txt
"""
}
process printHello2 {
tag "${str_in}"
input:
val str_in
output:
path("cheers.txt")
script:
"""
echo ${str_in.join(', ')} in Italian are ciao > cheers.txt
"""
}
/*
* A workflow can be named as a function and receive an input using the take keyword
*/
workflow first_pipeline {
take: str_input
main:
out = printHello(str_input)
emit: out
}
/*
* You can re-use the previous processes an combine as you prefer
*/
workflow second_pipeline {
take: str_input
main:
out = printHello2(str_input.collect())
emit: out
}
/*
* You can then invoke the different named workflows in this way
* passing the same input channel `str` to both
*/
workflow {
out1 = first_pipeline(str)
out2 = second_pipeline(str)
}
Change the pipeline to use only one process to handle both cases (either one element or a list).
You can choose the elements from a list using the positional keys (i.e. list[0], list[1], etc…).
Solution
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
str = Channel.from('hello', 'hola', 'bonjour')
process printHello {
tag { "${str_in}" }
input:
val str_in
output:
path("${str_in[0]}.txt")
script:
"""
echo ${str_in} in Italian is ciao > ${str_in[0]}.txt
"""
}
/*
* A workflow can be named as a function and receive an input using the take keyword
*/
workflow first_pipeline {
take: str_input
main:
out = printHello(str_input)
emit: out
}
/*
* You can re-use the previous processes an combine as you prefer
*/
workflow second_pipeline {
take: str_input
main:
out = printHello(str_input.collect())
emit: out
}
/*
* You can then invoke the different named workflows in this way
* passing the same input channel `str` to both
*/
workflow {
out1 = first_pipeline(str)
out2 = second_pipeline(str)
}
More complex scripts
We can feed the channel that is generated by a process to another process in the workflow definition. The variable used by AWK need to be escaped, otherwise they will be considered as proper Nextflow variables and thus produce an error. Every special character, e.g., $, needs to be escaped ($). It can be tedeous when writing long one liners; therefore, it is recommended to make a small shell script and call it as an executable. It has to be placed in a folder named bin inside the pipeline folder to be automatically considered from Nextflow as a tool in the path.
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
// the default "$baseDir/testdata/test.fa" can be overridden by using --inputfile OTHERFILENAME
params.inputfile = "$baseDir/testdata/test.fa"
// the "file method" returns a file system object given a file path string
sequences_file = file(params.inputfile)
// check if the file exists
if( !sequences_file.exists() ) exit 1, "Missing genome file: ${genome_file}"
/*
* Process 1 for splitting a fasta file in multiple files
*/
process splitSequences {
input:
path sequencesFile
output:
path ('seq_*')
// simple awk command
script:
"""
awk '/^>/{f="seq_"++d} {print > f}' < ${sequencesFile}
"""
}
/*
* Process 2 for reversing the sequences. Note the escaped AWK variables \$
*/
process reverseSequence {
tag { "${seq}" }
input:
path seq
output:
path "all.rev"
script:
"""
cat ${seq} | awk '{if (\$1~">") {print \$0} else system("echo " \$0 " |rev")}' > all.rev
"""
}
workflow {
splitted_seq = splitSequences(sequences_file)
// Here you have the output channel as a collection
splitted_seq.view()
// Here you have the same channel reshaped to send separately each value
splitted_seq.flatten().view()
// DLS2 allows you to reuse the channels! In past you had to create many identical
// channels for sending the same kind of data to different processes
rev_single_seq = reverseSequence(splitted_seq)
}
Here we have two simple processes:
the former splits the input fasta file into single sequences.
the latter is able to reverse the position of the sequences.
The input path is fed as a parameter using the script parameters ${seq}
params.inputfile
Note
The file “test.fa” is available in the github repository of the course
This value can be overridden when calling the script:
nextflow run test1.nf --inputfile another_input.fa
The workflow part connects the two processes so that the output of the first process becomes an input of the second process.
During the execution, Nextflow creates a number of temporary folders and also a soft link to the original input file. It will then store output files locally.
The output file is then linked in other folders for being used as input from other processes. This avoids clashes, and each process is isolated from the other.
nextflow run test1.nf -bg
N E X T F L O W ~ version 20.07.1
Launching `test1.nf` [sad_newton] - revision: 82e66714e4
[09/53e071] Submitted process > splitSequences
[/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_1, /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_2, /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_3]
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_1
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_2
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_3
[fe/0a8640] Submitted process > reverseSequence ([seq_1, seq_2, seq_3])
We can inspect the content of work/09/53e071* generated by the process splitSequences:
ls -l work/09/53e071*
total 24
-rw-r--r-- 1 lcozzuto staff 29 Oct 8 19:16 seq_1
-rw-r--r-- 1 lcozzuto staff 33 Oct 8 19:16 seq_2
-rw-r--r-- 1 lcozzuto staff 27 Oct 8 19:16 seq_3
lrwxr-xr-x 1 lcozzuto staff 69 Oct 8 19:16 test.fa -> /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/testdata/test.fa
File test.fa is a soft link to the original input.
If we inspect work/fe/0a8640* that is generated by the process reverseSequence, we see that the files generated by splitSequences are now linked as input.
ls -l work/fe/0a8640*
total 8
-rw-r--r-- 1 lcozzuto staff 89 Oct 8 19:16 all.rev
lrwxr-xr-x 1 lcozzuto staff 97 Oct 8 19:16 seq_1 -> /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_1
lrwxr-xr-x 1 lcozzuto staff 97 Oct 8 19:16 seq_2 -> /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_2
lrwxr-xr-x 1 lcozzuto staff 97 Oct 8 19:16 seq_3 -> /home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/09/53e071d286ed66f4020869c8977b59/seq_3
At this point we can make two different workflows to demonstrate how the new DSL allows reusing the code.
#!/usr/bin/env nextflow
nextflow.enable.dsl=2
// this can be overridden by using --inputfile OTHERFILENAME
params.inputfile = "$baseDir/testdata/test.fa"
// the "file method" returns a file system object given a file path string
sequences_file = file(params.inputfile)
// check if the file exists
if( !sequences_file.exists() ) exit 1, "Missing genome file: ${genome_file}"
/*
* Process 1 for splitting a fasta file in multiple files
*/
process splitSequences {
input:
path sequencesFile
output:
path ('seq_*')
// simple awk command
script:
"""
awk '/^>/{f="seq_"++d} {print > f}' < ${sequencesFile}
"""
}
/*
* Process 2 for reversing the sequences
*/
process reverseSequence {
tag { "${seq}" }
input:
path seq
output:
path "all.rev"
script:
"""
cat ${seq} | awk '{if (\$1~">") {print \$0} else system("echo " \$0 " |rev")}' > all.rev
"""
}
workflow flow1 {
take: sequences
main:
splitted_seq = splitSequences(sequences)
rev_single_seq = reverseSequence(splitted_seq)
}
workflow flow2 {
take: sequences
main:
splitted_seq = splitSequences(sequences).flatten()
rev_single_seq = reverseSequence(splitted_seq)
}
workflow {
flow1(sequences_file)
flow2(sequences_file)
}
The first workflow will just run like the previous script, while the second will “flatten” the output of the first process and will launch the second process on each single sequence.
The reverseSequence process of the second workflow will run in parallel if you have enough processors, or if you are running the script in a cluster environment, with a scheduler supported by Nextflow.
nextflow run test1.nf -bg
C02WX1XFHV2Q:nextflow lcozzuto$ N E X T F L O W ~ version 20.07.1
Launching `test1.nf` [insane_plateau] - revision: d33befe154
[bd/f4e9a6] Submitted process > flow1:splitSequences
[37/d790ab] Submitted process > flow2:splitSequences
[33/a6fc72] Submitted process > flow1:reverseSequence ([seq_1, seq_2, seq_3])
[87/54bfe8] Submitted process > flow2:reverseSequence (seq_2)
[45/86dd83] Submitted process > flow2:reverseSequence (seq_1)
[93/c7b1c6] Submitted process > flow2:reverseSequence (seq_3)
Directives
The directives are declaration blocks that can provide optional settings for a process.
For example, they can affect the way a process stages in and out the input and output files (stageInMode and stageOutMode), or they can indicate which file has to be considered a final result and in which folder it should be published (publishDir).
We can add the directive publishDir to our previous example:
/*
* Simple reverse the sequences
*/
process reverseSequence {
tag "$seq" // during the execution prints the indicated variable for follow-up
publishDir "output"
input:
path seq
output:
path "all.rev"
script:
"""
cat ${seq} | awk '{if (\$1~">") {print \$0} else system("echo " \$0 " |rev")}' > all.rev
"""
}
We can also use storeDir in case we want to have a permanent cache.
The process is executed only if the output files do not exist in the folder specified by storeDir.
When the output files exist, the process execution is skipped and these files are used as the actual process result.
We can also indicate what to do if a process fails.
The default is to stop the pipeline and to raise an error. But we can also skip the process using the errorStrategy directive:
errorStrategy 'ignore'
or retry a number of times changing the available memory or the maximum execution time, using the foolowing directives:
memory { 1.GB * task.attempt }
time { 1.hour * task.attempt }
errorStrategy 'retry'
maxRetries 3
Resuming a pipeline
You can resume the execution after the code modification using the parameter -resume.
nextflow run test1.nf -bg -resume
N E X T F L O W ~ version 20.07.1
Launching `test1.nf` [determined_celsius] - revision: eaf5b4d673
[bd/f4e9a6] Cached process > flow1:splitSequences
[37/d790ab] Cached process > flow2:splitSequences
[93/c7b1c6] Cached process > flow2:reverseSequence (seq_3)
[45/86dd83] Cached process > flow2:reverseSequence (seq_1)
[87/54bfe8] Cached process > flow2:reverseSequence (seq_2)
[33/a6fc72] Cached process > flow1:reverseSequence ([seq_1, seq_2, seq_3])
/home/ec2-user/git/CoursesCRG_Containers_Nextflow_May_2021/nextflow/nextflow/work/33/a6fc72786d042cacf733034d501691/all.rev
Note
IMPORTANT: Nextflow parameters are provided using one hyphen (-resume) while a pipeline parameters, two hyphens (--inputfile).
Sometimes you might want to resume a previous run of your pipeline.
To do so you need to extract the job id of that run. You can do this by using the command nextflow log.
nextflow log
TIMESTAMP DURATION RUN NAME STATUS REVISION ID SESSION ID COMMAND
2020-10-06 14:49:09 2s agitated_avogadro OK 61a595c5bf 4a7a8a4b-9bdb-4b15-9cc6-1b2cabe9a938 nextflow run test1.nf
2020-10-08 19:14:38 2.8s sick_edison OK 82e66714e4 4fabb863-2038-47b4-bac0-19e71f93f284 nextflow run test1.nf -bg
2020-10-08 19:16:03 3s sad_newton OK 82e66714e4 2d13e9f8-1ba6-422d-9087-5c6c9731a795 nextflow run test1.nf -bg
2020-10-08 19:30:59 2.3s disturbed_wozniak OK d33befe154 0a19b60d-d5fe-4a26-9e01-7a63d0a1d300 nextflow run test1.nf -bg
2020-10-08 19:35:52 2.5s insane_plateau OK d33befe154 b359f32c-254f-4271-95bb-6a91b281dc6d nextflow run test1.nf -bg
2020-10-08 19:56:30 2.8s determined_celsius OK eaf5b4d673 b359f32c-254f-4271-95bb-6a91b281dc6d nextflow run test1.nf -bg -resume
You can then resume the state of your execution using the SESSION ID:
nextflow run -resume 0a19b60d-d5fe-4a26-9e01-7a63d0a1d300 test1.nf
Nextflow’s cache can be disabled for a specific process by setting the directive cache to false. You can also choose among the three caching methods:
cache = true // (default) Cache keys are created indexing input files meta-data information (name, size and last update timestamp attributes).
cache = 'deep' // Cache keys are created indexing input files content.
cache = 'lenient' // (Best in HPC and shared file systems) Cache keys are created indexing input files path and size attributes
IMPORTANT: On some shared file systems you might have inconsistent file timestamps. Thus cache lenient prevents you from unwanted restarting of cached processes.
EXERCISE
Make the previous pipeline resilient to the process failing and save the results so the process execution would be skipped when the pipeline is launched again.
First, make the process reverseSequence to fail by introducing a typo in the command line, then add the directive to the process.
Solution
/*
* Broken process
*/
process reverseSequence {
tag { "${seq}" }
publishDir "output"
errorStrategy 'ignore'
input:
path seq
output:
path "all.rev"
script:
"""
cat ${seq} | AAAAAAA '{if (\$1~">") {print \$0} else system("echo " \$0 " |rev")}' > all.rev
"""
}
Write the first workflow using pipes. Nextflow DLS2 allows you to use pipes for connecting channels via input / output.
See the documentation on pipes.
Solution
workflow flow1 {
take: sequences
main:
splitSequences(sequences) | reverseSequence | view()
}