Input data and parameters
Input
| Analysis date: | Mon Mar 09 18:30:59 GMT 2026 |
| BAM file: | SRR3091420_1_chr6Aligned.sortedByCoord.out.bam |
| Counting algorithm: | uniquely-mapped-reads |
| GTF file: | /users/bi/lcozzuto/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf |
| Number of bases for 5'-3' bias computation: | 100 |
| Number of transcripts for 5'-3' bias computation: | 1,000 |
| Paired-end sequencing: | no |
| Protocol: | non-strand-specific |
| Sorting performed: | no |
Summary
Reads alignment
| Number of mapped reads: | 833,498 |
| Total number of alignments: | 886,156 |
| Number of secondary alignments: | 52,658 |
| Number of non-unique alignments: | 99,212 |
| Aligned to genes: | 688,876 |
| Ambiguous alignments: | 63,273 |
| No feature assigned: | 34,795 |
| Not aligned: | 0 |
| Strand specificity estimation (fwd/rev): | 0.52 / 0.48 |
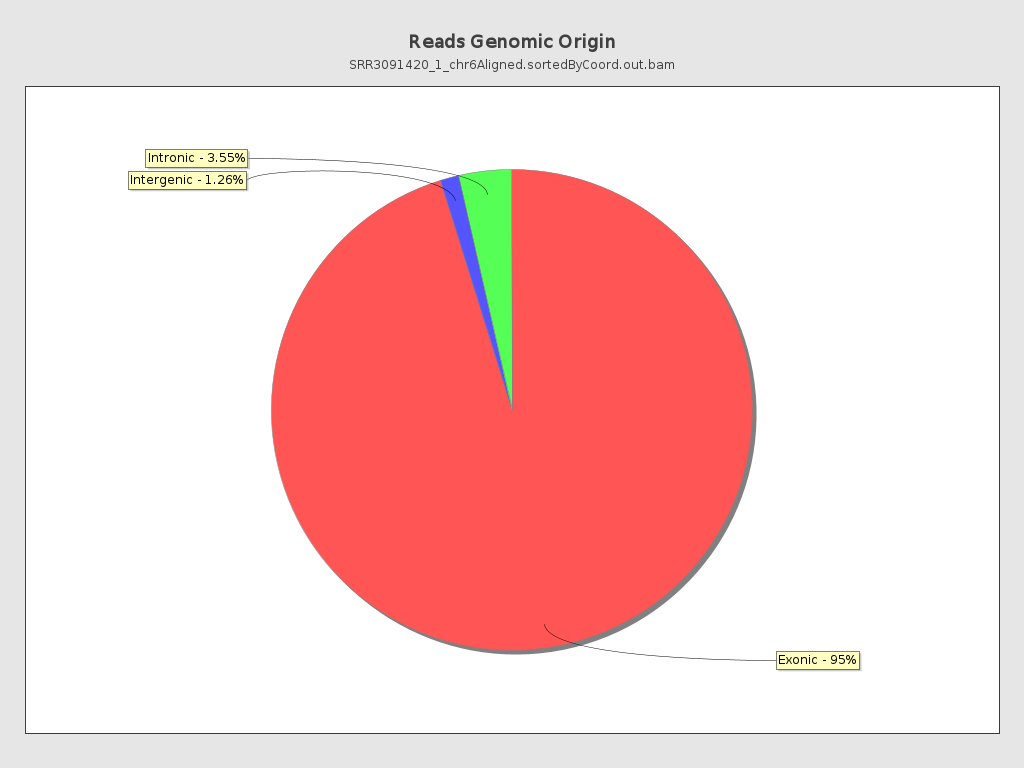
Reads genomic origin
| Exonic: | 688,876 / 95.19% |
| Intronic: | 25,684 / 3.55% |
| Intergenic: | 9,111 / 1.26% |
| Intronic/intergenic overlapping exon: | 17,970 / 2.48% |
Transcript coverage profile
| 5' bias: | 0.6 |
| 3' bias: | 0.36 |
| 5'-3' bias: | 1.78 |
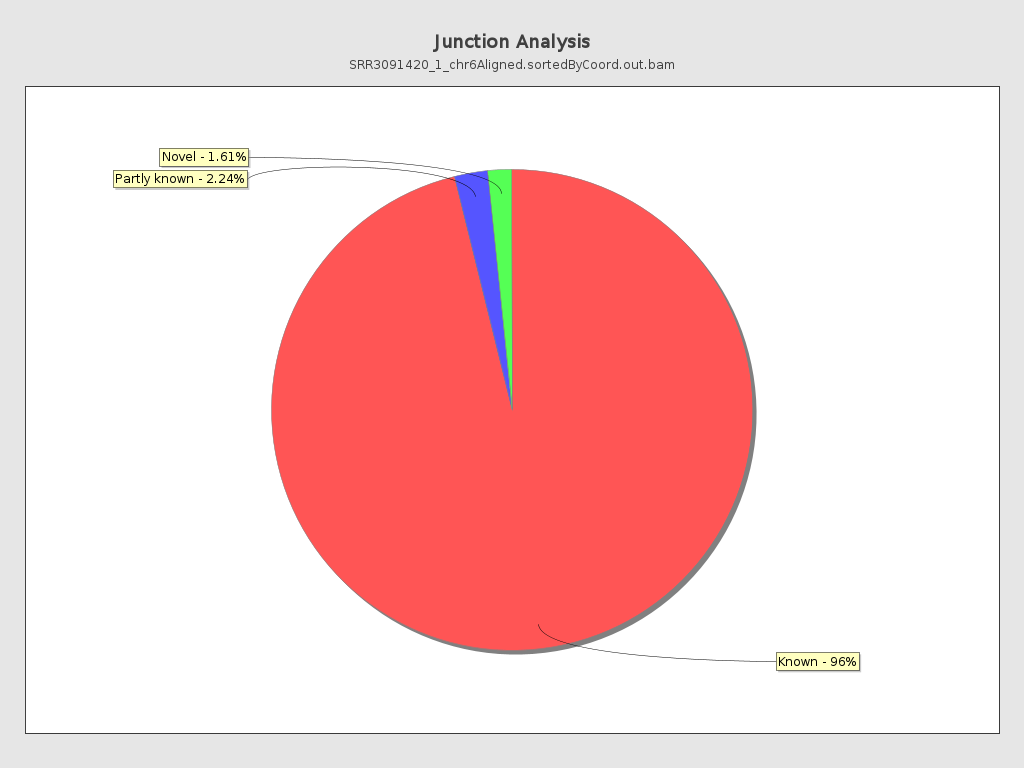
Junction analysis
| Reads at junctions: | 91,355 |
| ACCT | 16.31% |
| ATCT | 7.6% |
| AGGT | 5.91% |
| GCCT | 4.09% |
| ACCC | 3.55% |
| AGGC | 2.32% |
| AGAT | 2.24% |
| AGCA | 2.05% |
| TTCT | 1.85% |
| TCCT | 1.73% |
| AGCT | 1.69% |
.png)
.png)
.png)