GTF files
The General Transfer Format (GTF) format contains annotation of features and consists of one line per feature, each containing 9 columns of data.
GTF files can be downloaded for example from ENSEMBL, where each correspond to a specific version/update of the annotation.
The annotation from the latest version of the annotation is in the ENSEMBL main page (for Homo sapiens).
Previous versions of the annotation are found in the archives.
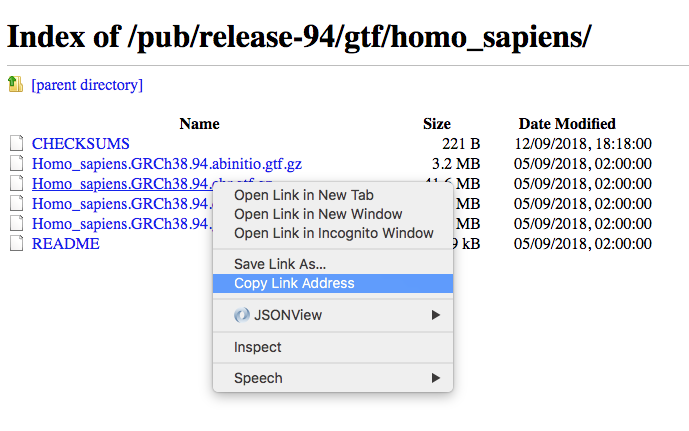
- Download the GTF file in ENSEMBL release 94 for Homo sapiens.
The name of the ENSEMBL gtf file is composed of:
- <species>: The systematic name of the species. (here, Homo_sapiens)
- <assembly>: The assembly build name. (here, GRCh38)
- <version>: The version of Ensembl from which the data was exported. (here, 94)
We can use this time the tool curl and the option -o for indicating the output file name.
curl -o Homo_sapiens.GRCh38.94.chr.gtf.gz ftp://ftp.ensembl.org/pub/release-94/gtf/homo_sapiens/Homo_sapiens.GRCh38.94.chr.gtf.gz
% Total % Received % Xferd Average Speed Time Time Time Current
Dload Upload Total Spent Left Speed
100 41.6M 100 41.6M 0 0 19.6M 0 0:00:02 0:00:02 --:--:-- 19.6M
- Let’s check the file:
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz | head
#!genome-build GRCh38.p12
#!genome-version GRCh38
#!genome-date 2013-12
#!genome-build-accession NCBI:GCA_000001405.27
#!genebuild-last-updated 2018-07
1 havana gene 11869 14409 . + . gene_id "ENSG00000223972"; gene_version "5"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene";
1 havana transcript 11869 14409 . + . gene_id "ENSG00000223972"; gene_version "5"; transcript_id "ENST00000456328"; transcript_version "2"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana"; transcript_biotype "processed_transcript"; tag "basic"; transcript_support_level "1";
1 havana exon 11869 12227 . + . gene_id "ENSG00000223972"; gene_version "5"; transcript_id "ENST00000456328"; transcript_version "2"; exon_number "1"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana"; transcript_biotype "processed_transcript"; exon_id "ENSE00002234944"; exon_version "1"; tag "basic"; transcript_support_level "1";
1 havana exon 12613 12721 . + . gene_id "ENSG00000223972"; gene_version "5"; transcript_id "ENST00000456328"; transcript_version "2"; exon_number "2"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana"; transcript_biotype "processed_transcript"; exon_id "ENSE00003582793"; exon_version "1"; tag "basic"; transcript_support_level "1";
1 havana exon 13221 14409 . + . gene_id "ENSG00000223972"; gene_version "5"; transcript_id "ENST00000456328"; transcript_version "2"; exon_number "3"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DDX11L1-202"; transcript_source "havana"; transcript_biotype "processed_transcript"; exon_id "ENSE00002312635"; exon_version "1"; tag "basic"; transcript_support_level "1";
The file is quite big. In general it has a header indicated by the first character ”#” and one row per feature composed by 9 columns:
| Column number | Column name | Details |
|---|---|---|
| 1 | seqname | name of the chromosome or scaffold; chromosome names can be given with or without the ‘chr’ prefix. |
| 2 | source | name of the program that generated this feature, or the data source (database or project name) |
| 3 | feature | feature type name, e.g. Gene, Variation, Similarity |
| 4 | start | Start position of the feature, with sequence numbering starting at 1. |
| 5 | end | End position of the feature, with sequence numbering starting at 1. |
| 6 | score | A floating point value. |
| 7 | strand | defined as + (forward) or - (reverse). |
| 8 | frame | One of ‘0’, ‘1’ or ‘2’. ‘0’ indicates that the first base of the feature is the first base of a codon, ‘1’ that the second base is the first base of a codon, and so on.. |
| 9 | attribute | A semicolon-separated list of tag-value pairs, providing additional information about each feature. |
Let’s count the number of gene features we have.
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz | awk '{if ($3=="gene") print }'| wc -l
58676
Exercise
Try to calculate the total number of each features. The result should be the following:
746300 CDS
1261907 exon
149893 five_prime_utr
58676 gene
120 Selenocysteine
86423 start_codon
78536 stop_codon
148456 three_prime_utr
206534 transcript
Now let’s focus on genes. We see in the description the following annotations: gene_id, gene_version, gene_name, gene_source and gene_biotype.
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz| awk '{if ($3=="gene") print}'| head -n 5
1 havana gene 11869 14409 . + . gene_id "ENSG00000223972"; gene_version "5"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene";
1 havana gene 14404 29570 . - . gene_id "ENSG00000227232"; gene_version "5"; gene_name "WASH7P"; gene_source "havana"; gene_biotype "unprocessed_pseudogene";
1 mirbase gene 17369 17436 . - . gene_id "ENSG00000278267"; gene_version "1"; gene_name "MIR6859-1"; gene_source "mirbase"; gene_biotype "miRNA";
1 havana gene 29554 31109 . + . gene_id "ENSG00000243485"; gene_version "5"; gene_name "MIR1302-2HG"; gene_source "havana"; gene_biotype "lincRNA";
1 mirbase gene 30366 30503 . + . gene_id "ENSG00000284332"; gene_version "1"; gene_name "MIR1302-2"; gene_source "mirbase"; gene_biotype "miRNA";
We can extract the 10th field of gene features by using awk but specifying this time the separator (tab). This to avoid to consider also space as possible separators.
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz| awk -F"\t" '{if ($3=="gene") print $9}'|head -n 5
gene_id "ENSG00000223972"; gene_version "5"; gene_name "DDX11L1"; gene_source "havana"; gene_biotype "transcribed_unprocessed_pseudogene";
gene_id "ENSG00000227232"; gene_version "5"; gene_name "WASH7P"; gene_source "havana"; gene_biotype "unprocessed_pseudogene";
gene_id "ENSG00000278267"; gene_version "1"; gene_name "MIR6859-1"; gene_source "mirbase"; gene_biotype "miRNA";
gene_id "ENSG00000243485"; gene_version "5"; gene_name "MIR1302-2HG"; gene_source "havana"; gene_biotype "lincRNA";
gene_id "ENSG00000284332"; gene_version "1"; gene_name "MIR1302-2"; gene_source "mirbase"; gene_biotype "miRNA";
We can then use either awk or sed to extract only the portion of text we are interested in
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz| awk -F"\t" '{if ($3=="gene") print $9}'| cut -d ";" -f 5 | cut -d " " -f 3 | head -n 5
"transcribed_unprocessed_pseudogene"
"unprocessed_pseudogene"
"miRNA"
"lincRNA"
"miRNA"
Now simply adding sort and uniq -c will allow us to get the statistics on biotypes.
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz| awk -F"\t" '{if ($3=="gene") print $9}'| cut -d ";" -f 5 | cut -d " " -f 3 |sort|uniq -c
32 "3prime_overlapping_ncRNA"
5583 "antisense"
73 "bidirectional_promoter_lncRNA"
14 "IG_C_gene"
9 "IG_C_pseudogene"
37 "IG_D_gene"
18 "IG_J_gene"
3 "IG_J_pseudogene"
1 "IG_pseudogene"
144 "IG_V_gene"
188 "IG_V_pseudogene"
7629 "lincRNA"
1 "macro_lncRNA"
1879 "miRNA"
2212 "misc_RNA"
2 "Mt_rRNA"
22 "Mt_tRNA"
2 "non_coding"
41 "polymorphic_pseudogene"
10189 "processed_pseudogene"
600 "processed_transcript"
19922 "protein_coding"
18 "pseudogene"
8 "ribozyme"
52 "rRNA"
499 "rRNA_pseudogene"
49 "scaRNA"
1 "scRNA"
894 "sense_intronic"
180 "sense_overlapping"
943 "snoRNA"
1900 "snRNA"
5 "sRNA"
1060 "TEC"
481 "transcribed_processed_pseudogene"
124 "transcribed_unitary_pseudogene"
884 "transcribed_unprocessed_pseudogene"
2 "translated_processed_pseudogene"
6 "TR_C_gene"
4 "TR_D_gene"
79 "TR_J_gene"
4 "TR_J_pseudogene"
106 "TR_V_gene"
33 "TR_V_pseudogene"
95 "unitary_pseudogene"
2647 "unprocessed_pseudogene"
1 "vaultRNA"
There are some characters in linux that are considered “special” such as dot, slash, backslash, quotes etc… because are interpreted as patterns. For avoiding this extra intepretation you need to escape them using backslash ().
So you can add to the previous example the code sed s/\"//g to remove the quotation marks too.
Exercise
- Let’s try to calculate the number of protein coding genes per chromosome. The results should be:
2043 1
1245 2
1076 3
752 4
884 5
1044 6
910 7
845 X
683 8
776 9
1309 11
729 10
1036 12
321 13
613 14
600 15
855 16
1186 17
269 18
543 20
1472 19
45 Y
442 22
231 21
13 MT
- Let’s try to calculate the number of exons per gene and to report also the gene name. We should redirect the result to a file since the numer of genes is about 60.000. The result should be:
9 "ENSG00000223972"; "DDX11L1";
11 "ENSG00000227232"; "WASH7P";
1 "ENSG00000278267"; "MIR6859-1";
5 "ENSG00000243485"; "MIR1302-2HG";
1 "ENSG00000284332"; "MIR1302-2";
5 "ENSG00000237613"; "FAM138A";
1 "ENSG00000268020"; "OR4G4P";
4 "ENSG00000240361"; "OR4G11P";
4 "ENSG00000186092"; "OR4F5";
17 "ENSG00000238009"; "AL627309.1";
...
- Create a BED file out of the genes from the GTF. TIPS awk allows you to change the column order! Solution
zcat Homo_sapiens.GRCh38.94.chr.gtf.gz | awk '$3=="gene" {OFS="\t"; print $1,$4,$5,".",$10,$7}' | sed s/\"//g|sed s/\;//g|head
1 11869 14409 . ENSG00000223972 +
1 14404 29570 . ENSG00000227232 -
1 17369 17436 . ENSG00000278267 -
1 29554 31109 . ENSG00000243485 +
1 30366 30503 . ENSG00000284332 +
1 34554 36081 . ENSG00000237613 -
1 52473 53312 . ENSG00000268020 +
1 57598 64116 . ENSG00000240361 +
1 65419 71585 . ENSG00000186092 +
1 89295 133723 . ENSG00000238009 -