4 Nextflow
Modules and how to re-use the code
A great advantage of the Nextflow is to allow the modularization of the code. In particular, you can move a named workflow within a module and keep it aside for being accessed by different pipelines.
The test4 folder provides an example of using modules.
/*
* NextFlow test pipe
* @authors
* Luca Cozzuto <lucacozzuto@gmail.com>
*
*/
/*
* Input parameters: read pairs
* Params are stored in the params.config file
*/
version = "1.0"
// this prevents a warning of undefined parameter
params.help = false
// this prints the input parameters
log.info """
BIOCORE@CRG - N F TESTPIPE ~ version ${version}
=============================================
reads : ${params.reads}
"""
// this prints the help in case you use --help parameter in the command line and it stops the pipeline
if (params.help) {
log.info 'This is the Biocore\'s NF test pipeline'
log.info 'Enjoy!'
log.info '\n'
exit 1
}
Channel
.fromPath( params.reads ) // read the files indicated by the wildcard
.ifEmpty { error "Cannot find any reads matching: ${params.reads}" } // if empty, complains
.set {reads_for_fastqc} // make the channel "reads_for_fastqc"
include { multiqc } from "${projectDir}/modules/multiqc"
include { fastqc } from "${projectDir}/modules/fastqc"
workflow {
fastqc_out = fastqc(reads_for_fastqc)
multiqc(fastqc_out.collect())
}
workflow.onComplete {
println ( workflow.success ? "\nDone! Open the following report in your browser --> ${multiqcOutputFolder}/multiqc_report.html\n" : "Oops .. something went wrong" )
}
We now include two modules, named fastqc and multiqc, from `${projectDir}/modules/fastqc.nf` and `${projectDir}/modules/multiqc.nf`.
Let’s inspect the multiQC module:
/*
* multiqc module
*/
process multiqc {
publishDir("multiqc_output", mode: 'copy')
input:
path (inputfiles)
output:
path "multiqc_report.html"
script:
"""
multiqc .
"""
}
The module multiqc takes as input a channel with files containing reads and produces as output the files generated by the multiqc program.
The module contains the directive publishDir, the tag, and the container to be used and has a similar input, output, and script session as the fastqc process in test3.nf.
A module can have hardcoded information such as the label and the container, but you can also specify some of them via nextflow.config.
In this example, we have the hardcoded labels, output director, and label:
/*
* fastqc module
*/
process fastqc {
tag { "${reads}" }
publishDir("fastqc_out", mode: 'copy')
container "quay.io/biocontainers/fastqc:0.11.9--0"
label 'onecpu'
input:
path(reads)
output:
path("*_fastqc*")
script:
"""
fastqc -t ${task.cpus} ${reads}
"""
}
In the multiqc module, we don’t specify the container. We indicate it via nextflow.config file using the withName selector.
Here you see that we are not using our own image, but rather we use the image provided by biocontainers in quay.
Note
IMPORTANT: You have to specify a default image to run nextflow -with-docker or -with-singularity and you have to have a container(s) defined inside modules.
Reporting and graphical interface
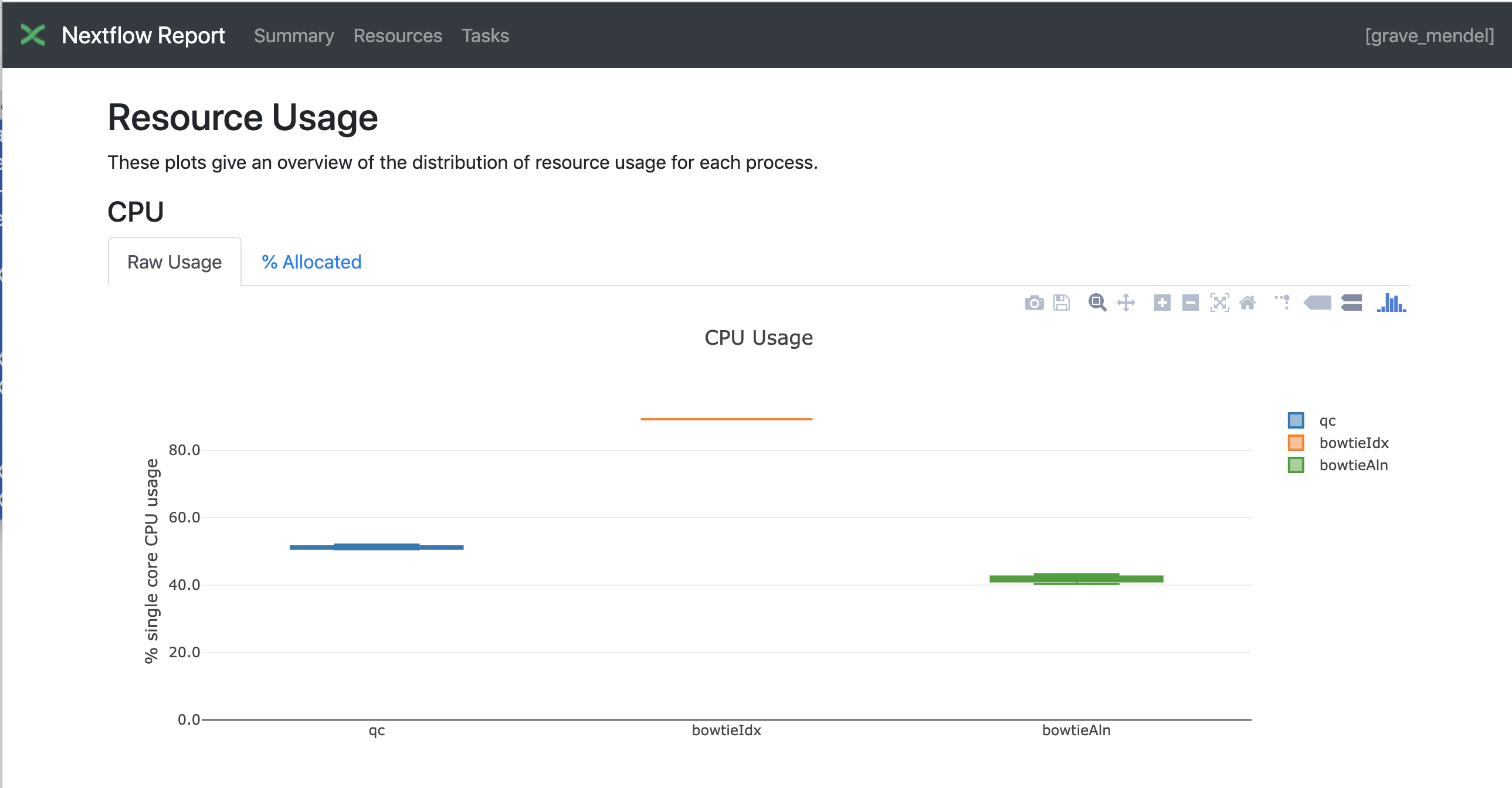
Nextflow has an embedded function for reporting information about the resources requested for each job and the timing; to generate a html report, run Nextflow with the -with-report parameter :
nextflow run test5.nf -with-docker -bg -with-report > log
Nextflow Seqera Platform (formerly known as Tower) is an open-source monitoring and management platform for Nextflow workflows. There are two versions:
Open source for monitoring of single pipelines.
Commercial one for workflow management, monitoring, and resource optimization.
We will show the open-source one.
First, you need to access the Seqera platform website and login.

We recommend using either Google or GitHub for login.
Once you are signed in you will see a page like this:
You can generate your token at ://cloud.seqera.io/tokens and copy-paste it into your pipeline using this snippet in the configuration file:
tower {
accessToken = '<YOUR TOKEN>'
enabled = true
}
or exporting those environmental variables:
export TOWER_ACCESS_TOKEN=*******YOUR***TOKEN*****HERE*******
Now we can launch the pipeline:
nextflow run test5.nf -with-singularity -with-tower -params-file params.yaml -bg > log
CAPSULE: Downloading dependency io.nextflow:nf-tower:jar:20.09.1-edge
CAPSULE: Downloading dependency org.codehaus.groovy:groovy-nio:jar:3.0.5
CAPSULE: Downloading dependency io.nextflow:nextflow:jar:20.09.1-edge
CAPSULE: Downloading dependency io.nextflow:nf-httpfs:jar:20.09.1-edge
CAPSULE: Downloading dependency org.codehaus.groovy:groovy-json:jar:3.0.5
CAPSULE: Downloading dependency org.codehaus.groovy:groovy:jar:3.0.5
CAPSULE: Downloading dependency io.nextflow:nf-amazon:jar:20.09.1-edge
CAPSULE: Downloading dependency org.codehaus.groovy:groovy-templates:jar:3.0.5
CAPSULE: Downloading dependency org.codehaus.groovy:groovy-xml:jar:3.0.5
and go to the tower website again:
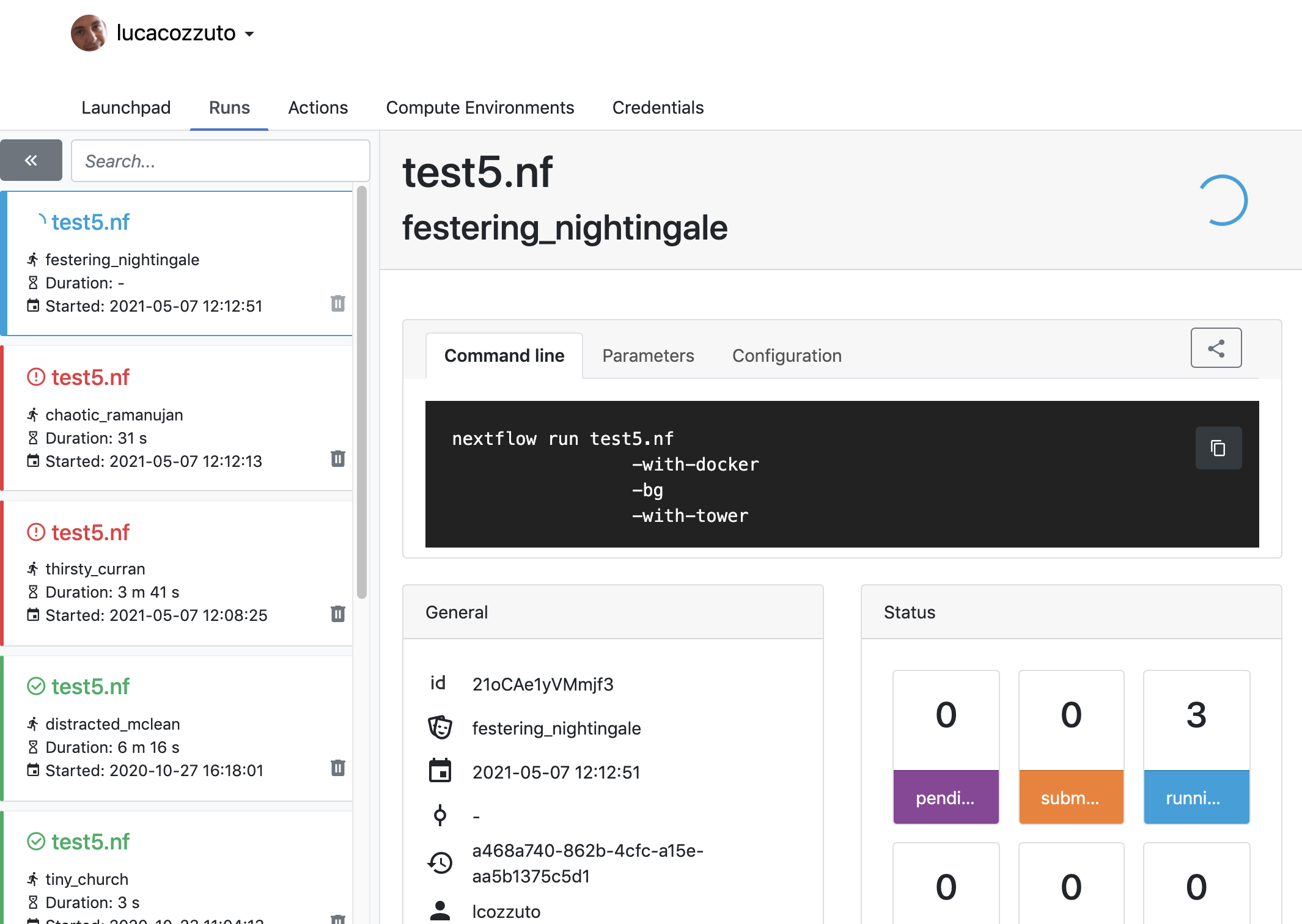
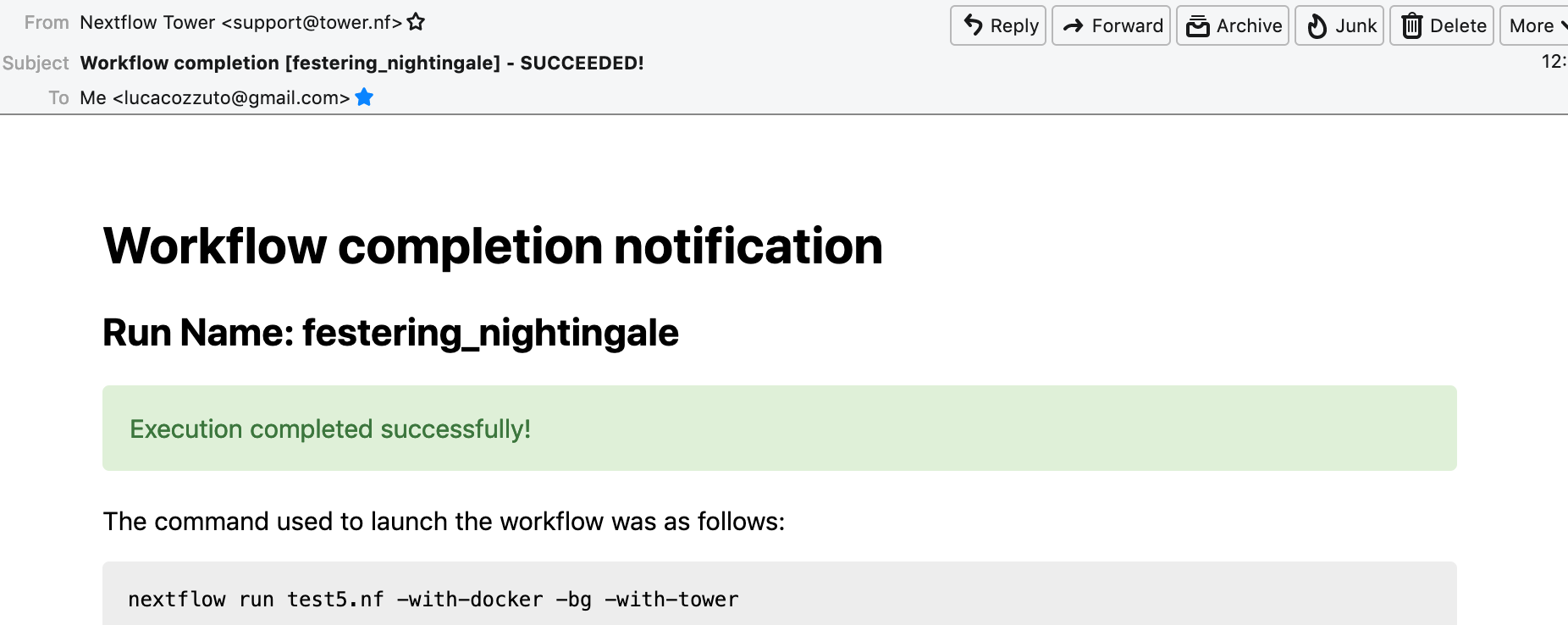
Once the pipeline is finished, you can also receive a mail.