

Read alignment to a reference genome
Once sequencing reads quality is ensured, we can proceed with their mapping (aka, alignment) to a reference genome. It will allow us to know to which regions of the reference genome most of the reads aligned to.
| Mapping of short reads |
|---|
 |
Before doing the mapping, we need to retrieve information about a reference genome from a public database.
The program which maps reads to a genome or transcriptome (called an aligner) needs a FASTA file of the reference genome (a file with an extension .fa).
Public resources on genome sequences and annotations
- GENCODE contains an accurate annotation of the human and mouse genes derived either using manual curation, computational analysis or targeted experimental approaches. GENCODE also contains information on functional elements, such as protein-coding loci with alternatively splices variants, non-coding loci and pseudogenes.
- Ensembl contains both automatically generated and manually curated annotations. They host different genomes and also comparative genomics data and variants. Ensembl genomes extends the genomic information across different taxonomic groups: bacteria, fungi, metazoa, plants, protists. Ensembl integrates also a genome browser.
- UCSC Genome Browser hosts information about different genomes. It integrates the GENCODE information as additional tracks.
We will use GENCODE for performing mapping to the human genome.
Let’s retrieve the data that we will use.
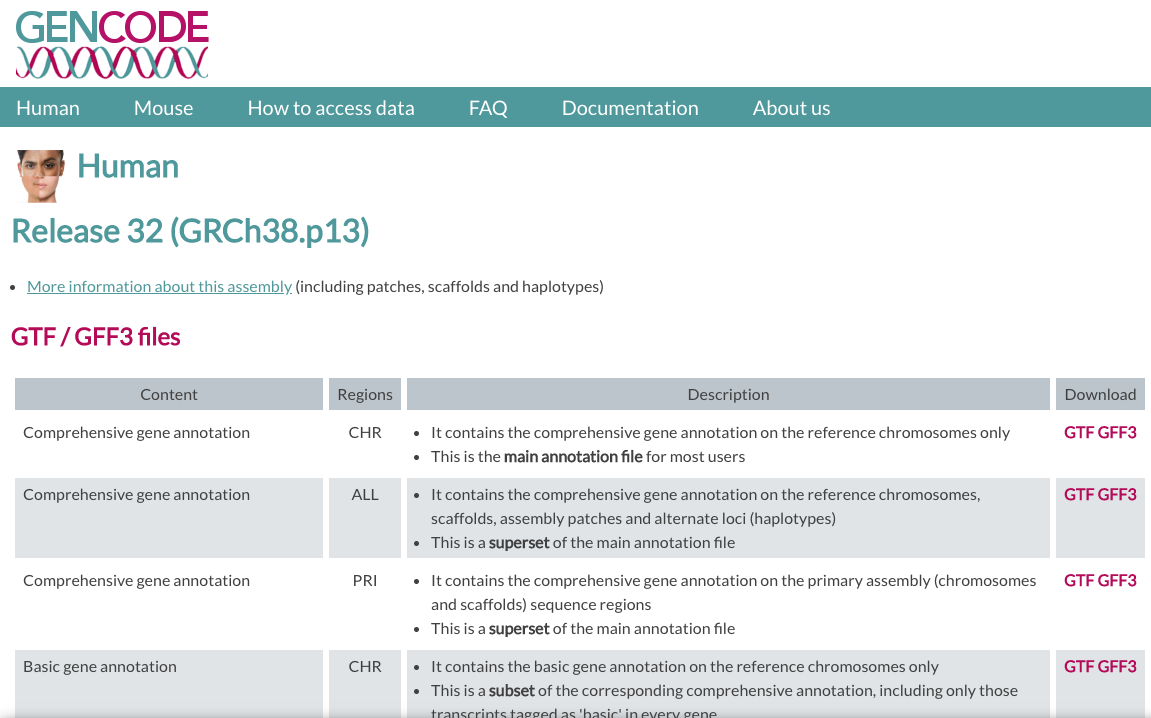
In GENCODE, we will be using the version v32 of the human genome.
We need two files:
- FASTA file for Genome sequence, primary assembly (GRCh38).
- GTF file for Comprehensive gene annotation (CHR)
| GENCODE website |
|---|
 |
FASTA formats
To speed up the mapping process, we already downloaded FASTA file for human v32 from GENCODE and extracted only the information about chromosome 21.
Download it to the folder db of your project:
pwd # to check where you are in the folder structure
cd ../ # to go to the root of the project folder, if needed
mkdir db
cd db
wget https://biocorecrg.github.io/PhD_course_genomics_format_2023/data/Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz
The genome is generally represented as a FASTA file (.fa file). Each chromosome sequence starts with a header row, starting with “>”:
zcat Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz | head -n 5
>chr21 dna:chromosome chromosome:GRCh38:21:1:46709983:1 REF
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
The size of the chromosome (46709983 bp) is already reported in the header, but we can check it using command wc.
Let’s first look at wc using our test file (created in the section “Fastq format”):
cat test
1
2
3
4
10
20
30
40
wc test
8 8 20 test
wc outputs 3 numbers: the number of lines, the number of words, and the number of characters.
zcat Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz | grep -v ">" | wc
778500 778500 47488483
Our combinations of commands is the following:
- unzipping the file to show the content to the stdout using zcat
- piping the output to the program grep for removing the part containing the header (check the -v option in grep, using man grep)
- counting the number of charachters using wc
Why the number of characters output by wc (47488483) is not equal to the number of base pairs (46709983)?
Note, the difference:
char=$(zcat Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz | grep -v ">" | wc -c)
echo $char
47488483
echo $((char-46709983))
778500
That tells us that every line of the file contains one additional character, which we can see using command cat -A (to show special characters):
zcat Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz | head -5 | cat -A
>21 dna:chromosome chromosome:GRCh38:21:1:46709983:1 REF$
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN$
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN$
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN$
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN$
This $ character stands for a new line character. How can we remove this character?
There is a Linux program tr that allows to manipulate the content of files in a command line. Let’s look at its options using man tr. Which option should we use to delete a character?
Let’s try it first with the test file:
cat test
1
2
3
4
10
20
30
40
cat test | wc
8 8 20
cat -A test
1$
2$
3$
4$
10$
20$
30$
40$
cat test | tr -d '\n'
123410203040
cat test | tr -d '\n' | wc
0 1 12
Now, we can apply these commands to our big file:
zcat Homo_sapiens.GRCh38.dna.chromosome.21.fa.gz | grep -v ">" | tr -d '\n' | wc -c
46709983
The flow of commands is the following:
- unzipping the file to show the content to the stdout using zcat
- piping the output to the program grep for removing the part containing the header (that starts with >)
- removing the carriage returns using tr
- counting the number of characters using wc
EXERCISE
Download in the folder ./db the annotation file at https://biocorecrg.github.io/PhD_course_genomics_format_2023/data/annotation.gtf.gz
- What is the date of this file? (use command more to explore the file)
- How many lines in this file?
- How many lines in this file contain the word “HAVANA”?
- How many characters the first line of the file contains?
- How many characters the lines starting with # contain?