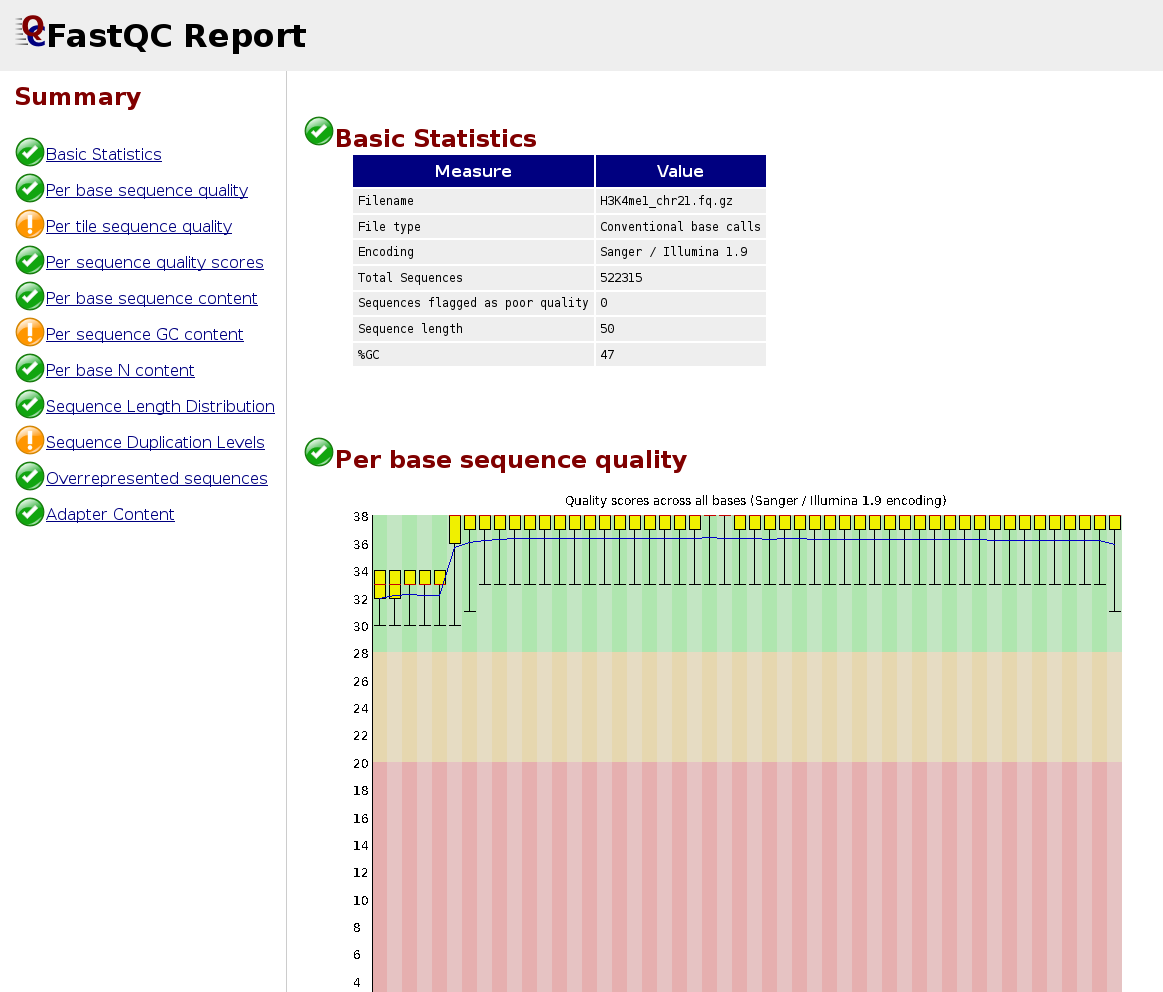
QC of sequencing reads
To assess the quality of sequencing data, we will use the program FastQC.
FastQC calculates statistics about the composition and quality of raw sequences (raw = as they come out of the sequencer).
FastQC outputs two files:
- H3K4me1_chr21_fastqc.html
- H3K4me1_chr21_fastqc.zip
Let’s create in the root of the project the folder fastqc, download there the html file and display the results in an Internet browser; e.g., Firefox, using command firefox:
cd .. # make sure to move up in the root of the project folder
mkdir fastqc
cd fastqc
wget https://biocorecrg.github.io/PhD_course_genomics_format_2024/data/H3K4me1_chr21_fastqc.html
firefox H3K4me1_chr21_fastqc.html
NOTE. If you don’t have X11 forwarding, don’t worry. Just copy paste this link in your browser:
https://biocorecrg.github.io/PhD_course_genomics_format_2024/data/H3K4me1_chr21_fastqc.html
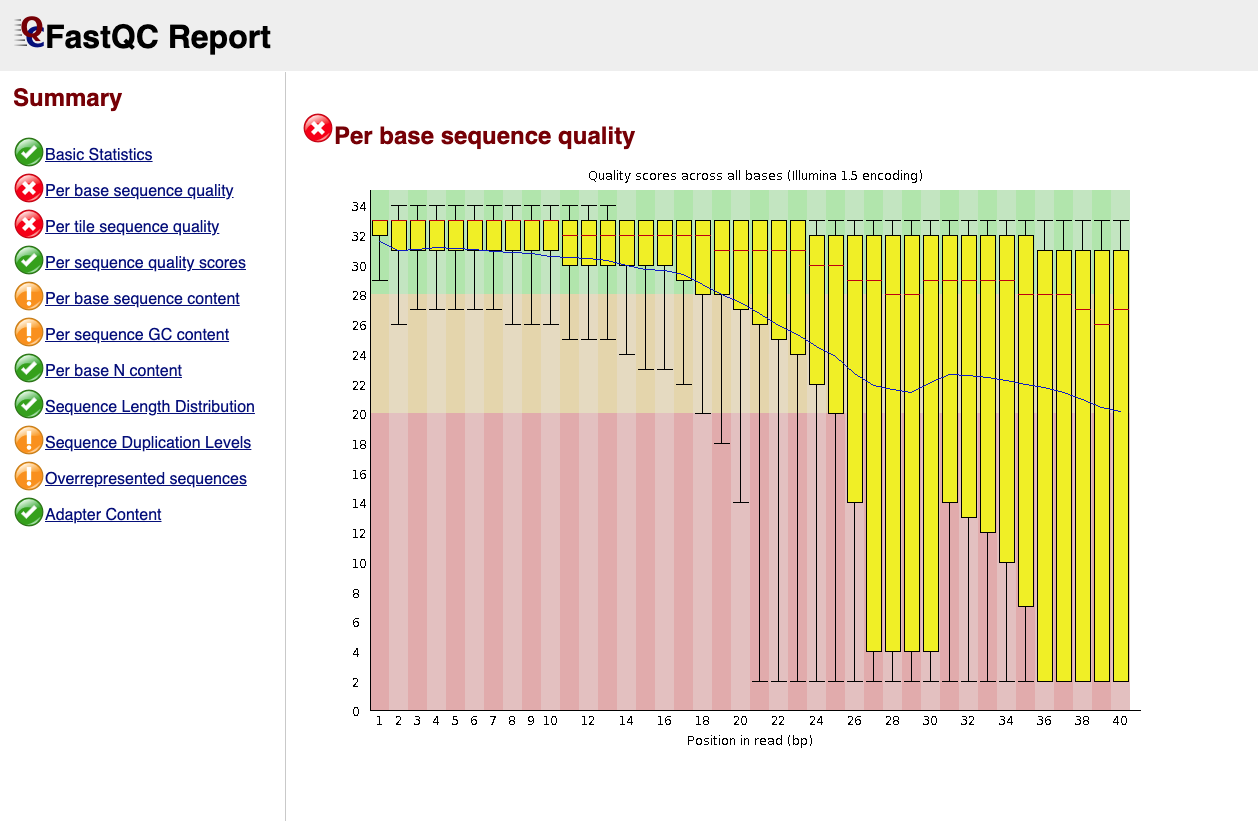
Below is an example of a poor quality dataset. As you can see, the average quality drops dramatically towards the 3’-end of sequencing reads.