MOP_DNA
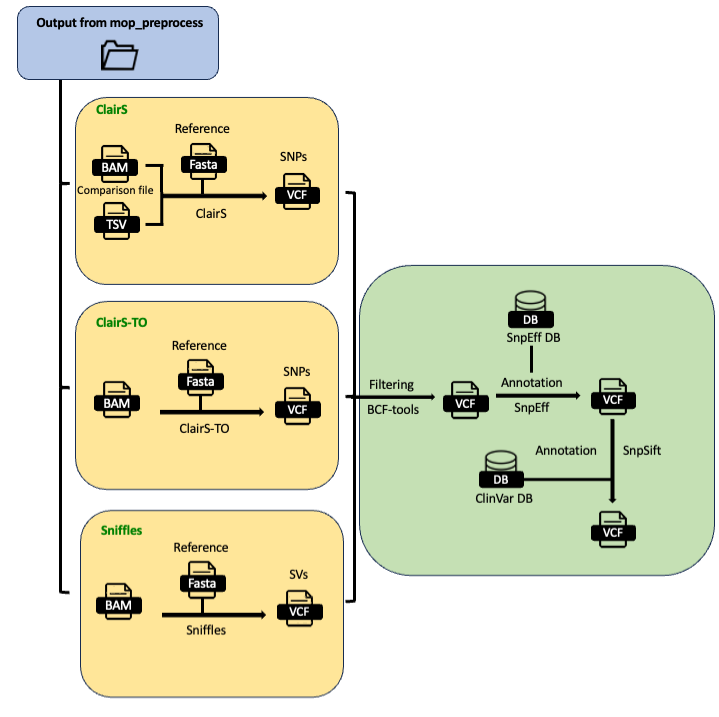
This pipeline takes as input the output from MOP_PREPROCESS and it outputs the SNPs and SVs together with their annotation.
Input Parameters
The input parameters are stored in yaml files like the one represented here:
input_path: "${projectDir}/../mop_preprocess/outfolder_mod"
##reference: "${projectDir}/GRCh38.primary_assembly.genome.fa"
reference: "../anno/curlcake_constructs.fasta.gz"
snpeff_db: "GRCh38.105"
clinvar_db: "/db/variants/clinvar/20240630/clinvar_20240630.vcf.gz"
clinvar_db_idx: "/db/variants/clinvar/20240630/clinvar_20240630.vcf.gz.tbi"
output: "${projectDir}/output_dna"
# clair flows
## clairS for comparing tumor/normal samples
clairS: "NO"
comparison: "${projectDir}/comparison.tsv"
## clairS-TO (tumor only)
clairS_TO: "NO"
## clair3
clair3: "YES"
## sniffles for structural variants
sniffles: "NO"
## Commmon flows
filter: "NO"
annotation: "NO"
slackhook: ""
email: ""
# Program params (workflows / tools)
progPars:
sniffles:
sniffles: "--tandem-repeats ${projectDir}/anno/human_GRCh38_no_alt_analysis_set.trf.bed"
clairS:
clairS: ""
clairS_TO:
clairS_TO: "--disable_indel_calling -p ont_r9_guppy"
clair3:
clair3: "--model_path=r1041_e82_260bps_fast_g632"
filter:
bcftools-view: "-f PASS"
common:
snpEff: ""
How to run the pipeline
Before launching the pipeline,user should:
Decide which containers to use - either docker or singularity [-with-docker / -with-singularity].
Fill in both params.config and tools_opt.tsv files.
To launch the pipeline, please use the following command:
nextflow run mop_dna.nf -params-file params.yaml -with-singularity > log.txt
You can run the pipeline in the background adding the nextflow parameter -bg:
nextflow run mop_dna.nf -params-file params.yaml -with-singularity -bg > log.txt
You can change the parameters either by changing params.config file or by feeding the parameters via command line:
nextflow run mop_dna.nf -params-file params.yaml -with-singularity -bg --output test2 > log.txt
You can specify a different working directory with temporary files:
nextflow run mop_dna.nf -params-file params.yaml -with-singularity -bg -w /path/working_directory > log.txt
Results
Several folders are created by the pipeline within the output directory specified by the output parameter:
clair_flow: contains the output of either clairS or clairS-TO.
sniffles_flow: contains the output of Sniffles tool.
filtered_vcf: contains the VCF produced previously that are being filtered.
annotation: contains the VCF produced previously together with annotations produced by SNPEff and ClinVar