MOP_MOD
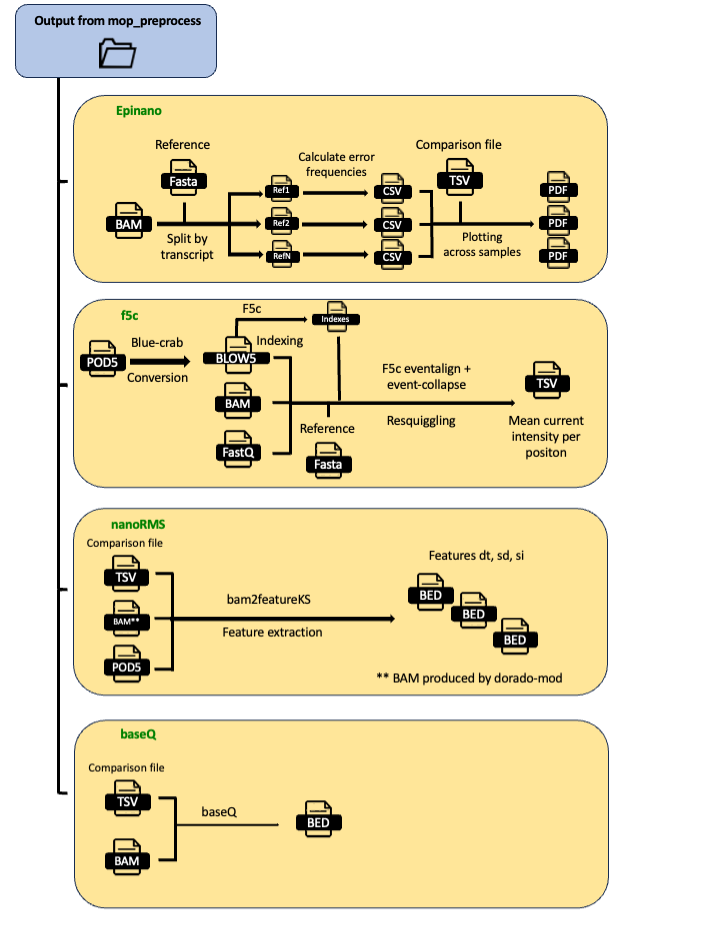
This pipeline takes as input the output from mop_preprocess and the pod5 that have been used as input. It runs 6 different workflows, 4 of which can be used later by mop_consensus: Epinano, F5c, nanoRMS, and baseQ. To use nanoRMS, you need to base the call with dorado-mod (i.e., with the option to emit the move table).
The remaining two are modKit and M6Anet:
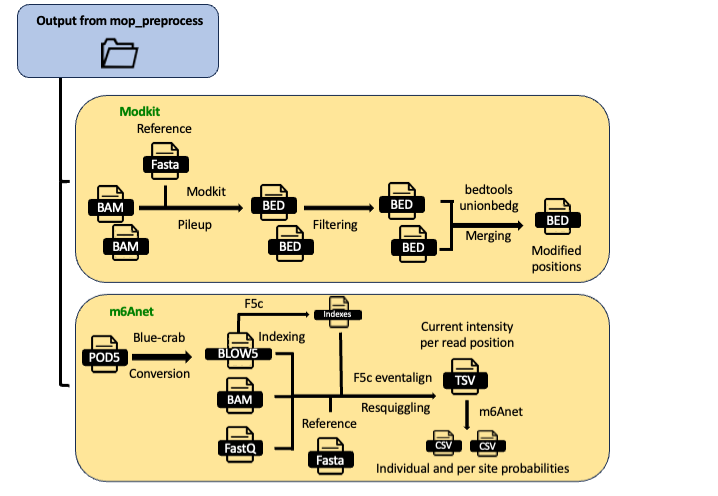
For running modkit, you need to basecall with dorado with models that include one or more modified bases.
Input Parameters
The input parameters are stored in yaml files like the one represented here:
input_path: "${projectDir}/../mop_preprocess/outfolder_mod/"
input_pod5: "${projectDir}/../data/pod5/dMOD/**/*.pod5"
comparison: "${projectDir}/comparison.tsv"
reference: "${projectDir}/../anno/curlcake_constructs.fasta.gz"
output: "${projectDir}/output_mod"
pars_tools: "${projectDir}/tools_opt.tsv"
# flows for nanoconsensus
epinano: "YES"
# nanoRMA needs mop_preprocess to be run with dorado-mod mode
nanoRMS: "YES"
baseQ: "YES"
f5c: "YES"
# Other flows
m6anet: "YES"
modkit: "YES"
#f5c kmer model
f5c_kmer_model: "${projectDir}/models/rna004.nucleotide.5mer.model"
# epinano plots
epinano_plots: "YES"
email: ""
# Program params (workflows / tools)
progPars:
epinano:
epinano: ""
f5c:
f5c: "--rna"
baseQ:
baseQ: ""
nanoRMS:
nanoRMS: ""
m6Anet:
f5c: "--rna"
inference: "--pretrained_model HEK293T_RNA004"
modkit:
modkit: "--mod-threshold 21891:0.90 --edge-filter 500,500"
cov_filtering: ""
strand: ""
field_sel: "11"
How to run the pipeline
Before launching the pipeline, user should:
Decide which containers to use - docker or singularity [-with-docker / -with-singularity].
Fill in both params.yaml and tools_opt.tsv files.
Fill in comparison.tsv file - please see example below:
wt_1 ko_1
wt_2 ko_2
To launch the pipeline, please use the following command:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity > log.txt
You can run the pipeline in the background adding the nextflow parameter -bg:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg > log.txt
You can change the parameters either by changing params.config file or by feeding the parameters via command line:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg --output test2 > log.txt
You can specify a different working directory with temporary files:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg -w /path/working_directory > log.txt
Note
If you have demultiplexed data in your mop_preprocess you need to turn on the option demulti_pod5: “ON”. The input of mop_mod will be:
input_pod5: "${projectDir}/../mop_preprocess/output/pod5_files/**/*.pod5"
Results
Several folders are created by the pipeline within the output directory specified by the output parameter:
Epinano
results are stored in epinano_flow directory. It contains two files per sample: one containing data at position level and the other, at 5-mer level. Different features frequencies as well as quality data are included in the results. See example below:
#Ref,pos,base,cov,q_mean,q_median,q_std,mis,ins,del
gene_A,2515,C,45497.0,5.36995,4.00000,3.97797,0.0822032221904741,0.18715519704595907,0.2058377475437941
gene_A,2516,A,45504.0,5.38207,4.00000,4.71619,0.17128164556962025,0.20497099156118143,0.07733386075949367
gene_A,2517,C,45529.0,6.92130,5.00000,5.04250,0.06165301236574491,0.1505633771881658,0.13540820136616222
gene_A,2518,A,45545.0,6.49821,5.00000,5.47485,0.10802503018992206,0.10855198155670216,0.2082775277198375
gene_A,2519,T,45557.0,6.51247,5.00000,4.81853,0.09386043857145993,0.14792457800118533,0.2033057488421099
To run this software, in the params.yaml file you should specify epinano: "YES".
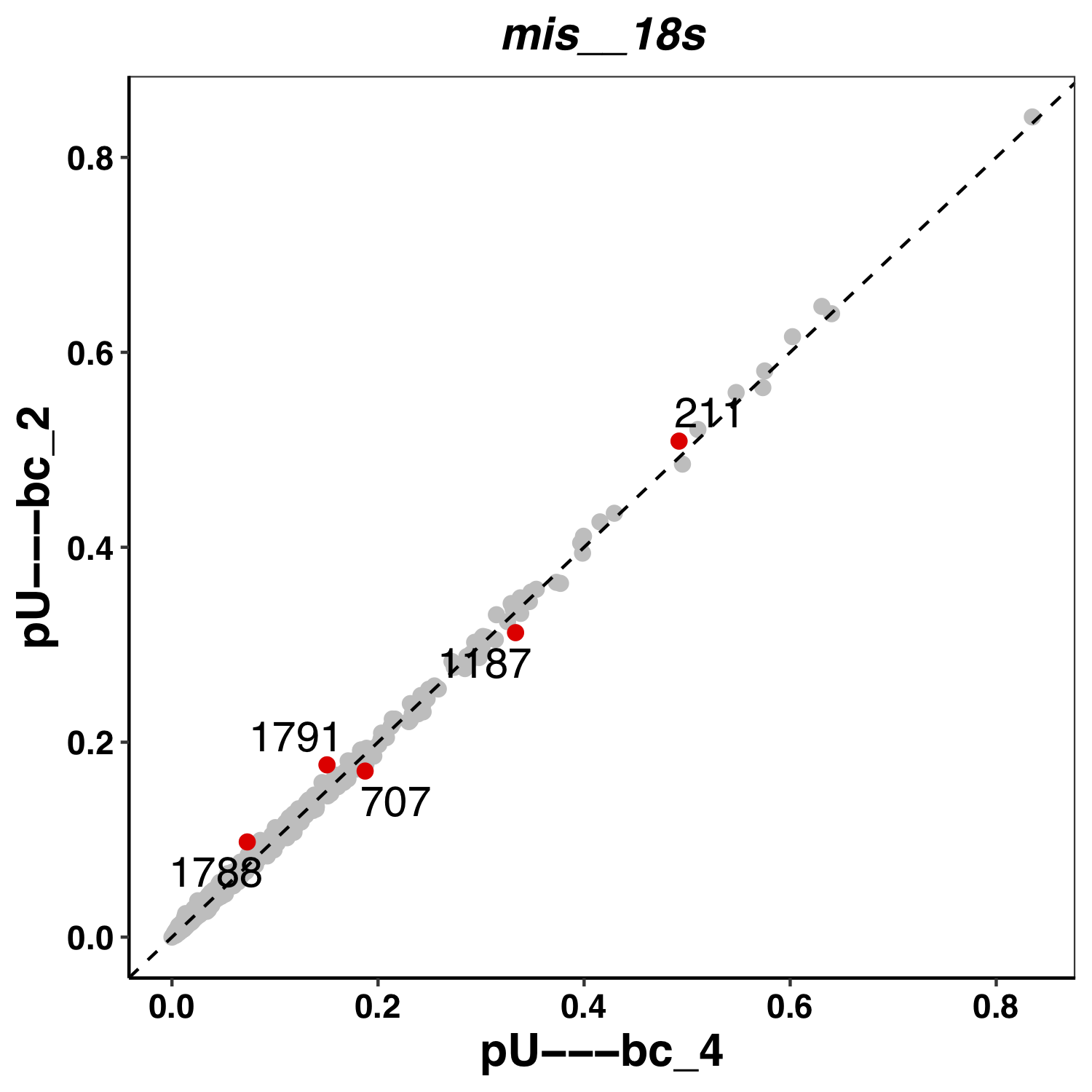
Here an example of a plot from Epinano:
F5c
F5c resquiggles the signal from the input pod5 using the alignments and the basecalled fastq files produced by mop_preprocess. It produces a compressed TSV file with mean current intensity per position.
To run this software, in the params.yaml file you should specify f5c: "YES".
nanoRMS
nanoRMS uses the move table stored in the bam file when basecalling with dorado-mod in mop_preprocess for predicting the modified bases. It needs a comparison file (unmodified vs modified) and it generates three BED files per comparison.
To run this software, in the params.yaml file you should specify nanoRMS: "YES".
baseQ
baseQ uses just the bam files for predicting the modified bases. It needs a comparison file (unmodified vs modified) and it generates a BED file per comparison.
To run this software, in the params.yaml file you should specify baseQ: "YES".
Modkit
Modkit is used for extracting modification encoded within BAM files (basecalling with modification)
Once the data has been basecalled with a modification-aware basecalling model, modification can be evaluated using modkit.
To run this software, in the params.yaml file you should specify modkit: "YES".
Modkit will generate a pileup file that can be filtered by keeping only the positions with a coverage higher than a value indicated in params.yaml. Moreover, you can select the strand and which field to be reported. Default is 11 (percent_modified). Here the list of fields:
chrom, chromStart, chromEnd, name, score, strand, thickStart, thickEnd, color, valid_coverage, percent_modified, count_modified, count_canonical, count_other_mode, count_delete, count_fail, count_diff, count_nocall
modkit:
modkit: "--mod-threshold 21891:0.90 --edge-filter 500,500"
cov_filtering: ""
strand: ""
field_sel: "11"
The resulting bedgraphs are then merged in a single table called union.bed.
m6Anet
The m6Anet workflow uses f5c for resquiggling the signal stored in the POD5 files using alignments in BAM and basecalled reads in FASTQ generated by mop_preprocess. The current intensity per read position is then used by m6Anet for m6A prediction. The results is stored in a folder per sample and it consists of two CSV files, one for individual probabilities and one for site probabilities.
To run this software, in the params.yaml file you should specify m6Anet: "YES".