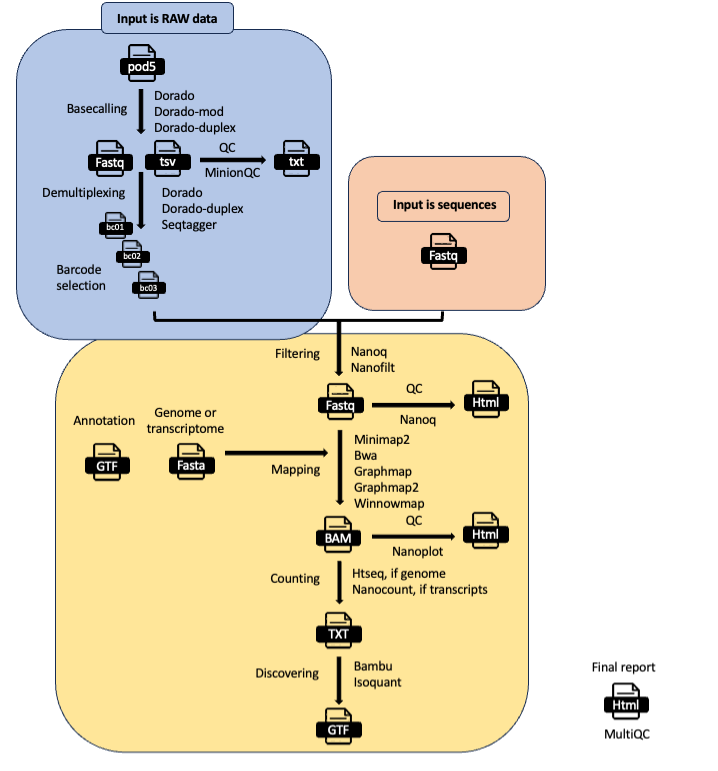
MOP_PREPROCESS
This pipeline takes as input the raw reads - single or multi - and it produces several outputs (basecalled pod5, sequences in fastq format, aligned reads in BAM format etc). The pre-processing pipeline can perform base-calling, demultiplexing (optional), filtering, quality control, mapping to a reference (either a genome or a transcriptome), feature counting, discovery of novel transcripts, and it generates a final report with the performance and results of each of the steps performed.
It support the new pod5 format. The basecalling can be performed with dorado or dorado-duplex and the demultiplexing with either dorado or seqtagger. Basecalled fastq and pod5 files can be demultiplexed as well. You can restrict the number of barcodes by indicating a file with barcode list using the barcodes parameter.
Input Parameters
The input parameters are stored in yaml files like the one represented here:
# Parameters
# Needed for pod5 input
pod5: ""
## Can be OFF / cuda10 / cuda11.
GPU: "OFF"
# Basecalling can be either NO, dorado, dorado-mod or dorado-duplex
basecalling: "NO"
#For emitting the move tables (with dorado-mod, can be ON / YES or empty)
emit_moves: ""
## Demultiplexing can be either dorado (for DNA) / seqtagger (for RNA)
#demultiplexing: "seqtagger"
demultiplexing: "dorado"
demulti_pod5: "ON"
### Number of fast5 basecalled per parallel job
granularity: 1
### File with the list of accepted barcodes. It can be empty
barcodes: ""
# Needed for fastq input
fastq: "${projectDir}/../data/fastq/*.fq.gz"
# Common
reference: "${projectDir}/../anno/yeast_rRNA_ref.fa.gz"
## Can be transcriptome / genome
ref_type: "transcriptome"
annotation: ""
## Can be nanoq / nanofilt
filtering: "nanoq"
## Can be graphmap / graphmap2 / minimap2 / winnowmap / bwa / NO
mapping: "minimap2"
## Can be nanocount for transcriptome / htseq for genome
counting: "nanocount"
## Can be NO / bambu / isoquant
discovery: "NO"
## Convert bam to cram
cram_conv: "YES"
subsampling_cram: 50
## Output and messaging
slackhook: ""
email: ""
output: "./outfolder/"
# Program params
progPars:
basecalling:
dorado: "sup"
dorado-duplex: "sup"
dorado-mod: "sup,m6A_DRACH"
demultiplexing:
seqtagger: "-k b100"
dorado: ""
filtering:
seqkit: ""
nanoq: ""
mapping:
graphmap: ""
graphmap2: "-x rnaseq"
minimap2: "-y -uf -ax splice -k14"
bwa: ""
winnowmap: ""
counting:
htseq: "-a 0"
nanocount: ""
discovery:
bambu: ""
You can change them by editing this file or using the command line as explained in the next section.
How to run the pipeline
Before launching the pipeline, the user can decide which containers to use: either docker or singularity [-with-docker / -with-singularity].
Then, to launch the pipeline, please use the following command by specifying the path of the yaml parameter file:
nextflow run mop_preprocess.nf -with-singularity -params-file params.yaml > log.txt
You can run the pipeline in the background by adding the nextflow parameter -bg:
nextflow run mop_preprocess.nf -params-file params.yaml -with-singularity -bg > log.txt
You can change the parameters either by changing the yaml config file or by feeding the parameters via command line:
nextflow run mop_preprocess.nf -with-singularity -params-file params.yaml -bg --output test2 > log.txt
You can specify a different working directory with temporary files:
nextflow run mop_preprocess.nf -with-singularity -params-file params.yaml -bg -w /path/working_directory > log.txt
You can use different profiles for running the pipeline in different environments. We have one set up for HPC using the SGE scheduler:
nextflow run mop_preprocess.nf -with-singularity -bg -params-file params.yaml -w /path/working_directory -profile cluster > log.txt
One for HPC using the slurm scheduler
nextflow run mop_preprocess.nf -with-singularity -bg -params-file params.yaml -w /path/working_directory -profile slurm > log.txt
One for emulating the new M1 processor for Apple:
nextflow run mop_preprocess.nf -with-singularity -bg -params-file params.yaml -w /path/working_directory -profile m1mac > log.txt
or you can run the pipeline locally:
nextflow run mop_preprocess.nf -with-singularity -bg -params-file params.yaml -w /path/working_directory -profile local > log.txt
Note
In case of errors you can troubleshoot by seeing the log file (log.txt) for more details. Furthermore, if more information is needed, you can also go to the intermediate directory indicated in the log and check both the .command.log and .command.err files.
Tip
Once the error has been solved or if you change a specific parameter, you can resume the execution with the Netxtlow parameter - resume (only one dash!). If there is an error, the pipeline will resume from the process that had the error and proceed with the rest. If you change a parameter, only the processes affected by this parameter will be re-run.
----------------------CHECK TOOLS -----------------------------
basecalling : dorado
> demultiplexing will be skipped
mapping : minimap2
filtering : nanoq
counting : nanocount
> discovery will be skipped
--------------------------------------------------------------
Skipping the email
[15/cc7992] Cached process > checkRef (Checking curlcake_constructs.fasta.gz)
[e7/fe0c2b] Submitted process > BASECALL:DORADO_BASECALL:downloadModel (A---1)
[a8/fc6e07] Submitted process > BASECALL:DORADO_BASECALL:baseCallMod (A---1)
[95/044f06] Submitted process > BASECALL:DORADO_BASECALL:baseCallMod (m6A---2)
[a1/867fa2] Submitted process > BASECALL:DORADO_BASECALL:bam2ModFastq (A---1)
Note
To resume the execution, temporary files generated previously by the pipeline must be kept. Otherwise, the pipeline will re-start from the beginning.
Command line options
The command line options for each tool used in the pipeline are stored within in the same yaml file with other parameters. The section is called progPars. Here is an example:
# Parameters
# Needed for pod5 input
pod5: ""
## Can be OFF / cuda10 / cuda11.
GPU: "OFF"
# Basecalling can be either NO, dorado, dorado-mod or dorado-duplex
basecalling: "NO"
#For emitting the move tables (with dorado-mod, can be ON / YES or empty)
emit_moves: ""
## Demultiplexing can be either dorado (for DNA) / seqtagger (for RNA)
#demultiplexing: "seqtagger"
demultiplexing: "dorado"
demulti_pod5: "ON"
### Number of fast5 basecalled per parallel job
granularity: 1
### File with the list of accepted barcodes. It can be empty
barcodes: ""
# Needed for fastq input
fastq: "${projectDir}/../data/fastq/*.fq.gz"
# Common
reference: "${projectDir}/../anno/yeast_rRNA_ref.fa.gz"
## Can be transcriptome / genome
ref_type: "transcriptome"
annotation: ""
## Can be nanoq / nanofilt
filtering: "nanoq"
## Can be graphmap / graphmap2 / minimap2 / winnowmap / bwa / NO
mapping: "minimap2"
## Can be nanocount for transcriptome / htseq for genome
counting: "nanocount"
## Can be NO / bambu / isoquant
discovery: "NO"
## Convert bam to cram
cram_conv: "YES"
subsampling_cram: 50
## Output and messaging
slackhook: ""
email: ""
output: "./outfolder/"
# Program params
progPars:
basecalling:
dorado: "sup"
dorado-duplex: "sup"
dorado-mod: "sup,m6A_DRACH"
demultiplexing:
seqtagger: "-k b100"
dorado: ""
filtering:
seqkit: ""
nanoq: ""
mapping:
graphmap: ""
graphmap2: "-x rnaseq"
minimap2: "-y -uf -ax splice -k14"
bwa: ""
winnowmap: ""
counting:
htseq: "-a 0"
nanocount: ""
discovery:
bambu: ""
The second level indicates the processing step as basecalling or demultiplexing etc, while the third indicates the tool. Finally you have the command specific command line between quotation marks.
Note
You can indicate the models to be used for basecalling with dorado or dorardo-duplex as “sup,m6A_DRACH”. The pipeline will try to download before and then to perform the basecalling. In case you want a specific model version you need to indicate the base simplex model as “rna002_70bps_hac@v3,pseU”. You can see here the list of models and modifications.
Tip
You don’t need to specify the whole path for the models of seqtagger, just the name of the model will be enough
Model libraries for specific tools
The following folders are available for the respective tools. Some models are already pre-installed-
- seqtagger_models
b04_RNA002
b04_RNA004
You also need to add the dedicated parameter within the tool_opts file for the specific tool as:
Note
You need to copy the model in the corresponding folder and indicate just the model name. You don’t need the absolute path.
Barcodes
You can select the barcodes you are interested in by writing them down in a text file as in this example. The format is samplename—barcodeID
lserrano---barcode01
lserrano---barcode02
lserrano---barcode03
lserrano---unclassified
The sample id is given by either the folder containing the fast5 files or the basename of the fastq files. So, if your files are in a folder named myfiles, it will be:
myfiles---bc_1
myfiles---bc_2
myfiles---bc_3
Note
The naming convention of the different barcodes is decided by each tool, so seqtagger will produce bc_1, bc_2, etc. while guppy will produce barcode01, barcode02, etc.
PolyA tail prediction
You can add the dorado parameters to predict polyA tail in the params.yaml file.
# Program params
progPars:
basecalling:
dorado: "sup --estimate-poly-a"
dorado-duplex: "sup --estimate-poly-a"
dorado-mod: "sup,m6A_DRACH --estimate-poly-a"
Results
Several folders are created by the pipeline within the output directory specified by the output parameter:
pod5_files: Contains the basecalled multifast5 files. Each batch can contain a variable number of sequences.
fastq_files: Contains one or, in case of demultiplexing, more fastq files.
QC_files: Contains each single QC produced by the pipeline.
alignment: Contains the bam file(s).
cram_files: Contains the cram file(s).
counts: Contains read counts per gene / transcript if counting was performed.
assigned: Contains assignment of each read to a given gene / transcript if counting was performed.
report: Contains the final multiqc report.
assembly: It contains assembled transcripts.