MOP_MOD
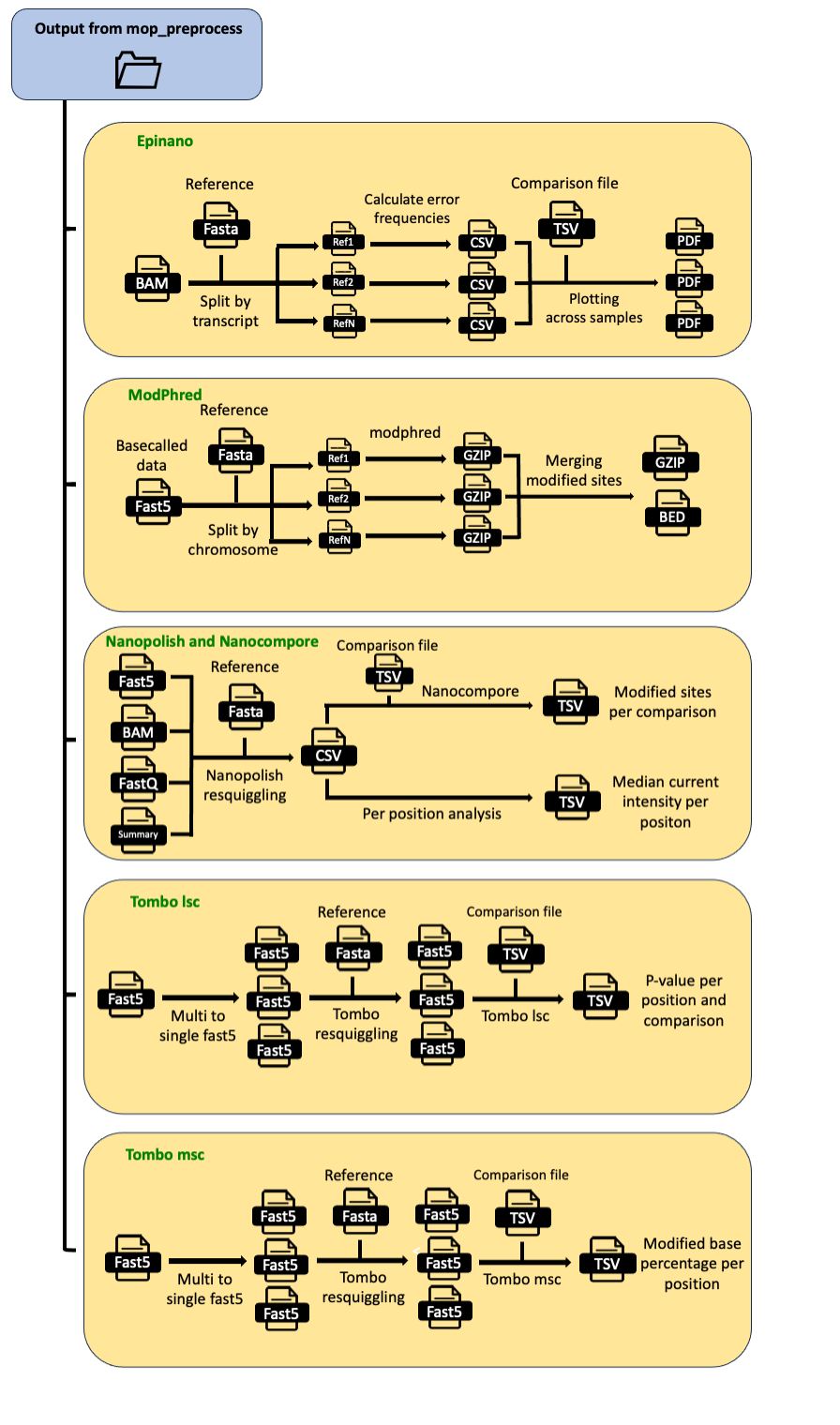
This pipeline takes as input the output from MOP_PREPROCESS: basecalled fast5 reads, together with their respective fastq files and unspliced alignments to the transcriptome . It runs four different RNA detection algorithms (Epinano, Nanopolish, Tombo and Nanocompore) and it outputs the predictions generated by each one of them as individual tab-delimited files.
Input Parameters
The input parameters are stored in yaml files like the one represented here:
input_path: "${projectDir}/../mop_preprocess/outfolder/"
comparison: "${projectDir}/comparison.tsv"
reference: "${projectDir}/../anno/yeast_rRNA_ref.fa.gz"
output: "${projectDir}/output_mod"
pars_tools: "${projectDir}/tools_opt.tsv"
# flows
epinano: "YES"
nanocompore: "NO"
tombo_lsc: "YES"
tombo_msc: "YES"
modphred: "NO"
# epinano plots
epinano_plots: "YES"
email: ""
How to run the pipeline
Before launching the pipeline, user should:
Decide which containers to use - either docker or singularity [-with-docker / -with-singularity].
Fill in both params.yaml and tools_opt.tsv files.
Fill in comparison.tsv file - please see example below:
wt_1 ko_1
wt_2 ko_2
To launch the pipeline, please use the following command:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity > log.txt
You can run the pipeline in the background adding the nextflow parameter -bg:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg > log.txt
You can change the parameters either by changing params.config file or by feeding the parameters via command line:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg --output test2 > log.txt
You can specify a different working directory with temporary files:
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg -w /path/working_directory > log.txt
Results
Several folders are created by the pipeline within the output directory specified by the output parameter:
Epinano results are stored in epinano_flow directory. It contains two files per sample: one containing data at position level and the other, at 5-mer level. Different features frequencies as well as quality data are included in the results. See example below:
#Ref,pos,base,cov,q_mean,q_median,q_std,mis,ins,del
gene_A,2515,C,45497.0,5.36995,4.00000,3.97797,0.0822032221904741,0.18715519704595907,0.2058377475437941
gene_A,2516,A,45504.0,5.38207,4.00000,4.71619,0.17128164556962025,0.20497099156118143,0.07733386075949367
gene_A,2517,C,45529.0,6.92130,5.00000,5.04250,0.06165301236574491,0.1505633771881658,0.13540820136616222
gene_A,2518,A,45545.0,6.49821,5.00000,5.47485,0.10802503018992206,0.10855198155670216,0.2082775277198375
gene_A,2519,T,45557.0,6.51247,5.00000,4.81853,0.09386043857145993,0.14792457800118533,0.2033057488421099
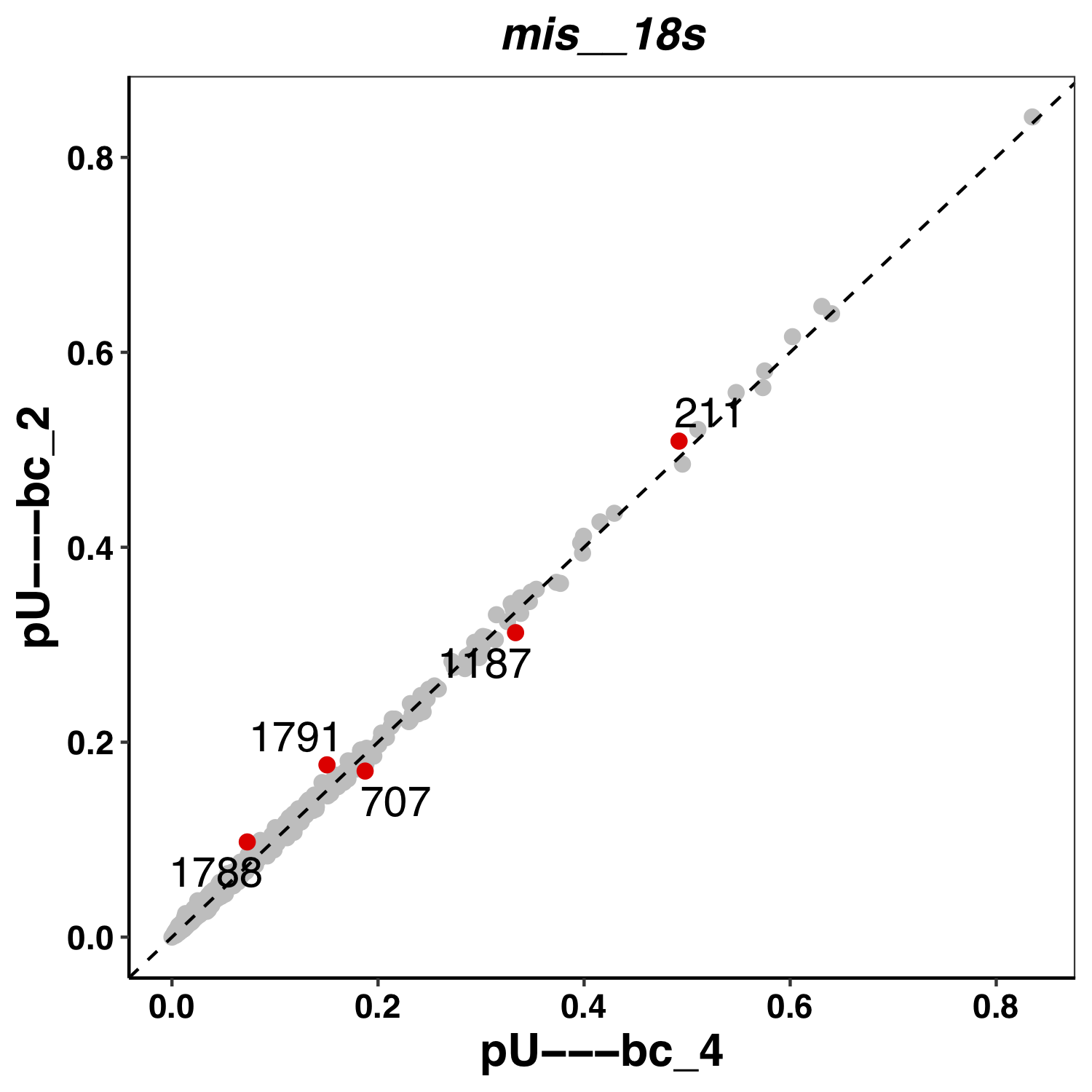
Here an example of a plot from Epinano:
Tombo results are stored in tombo_flow directory. It contains one file per comparison. It reports the p-value per position, the sum of p-values per 5-mer and coverage in both WT and KO. See example below:
"Ref_Position" "Chr" "Position" "Tombo_SiteScore" "Coverage_Sample" "Coverage_IVT" "Tombo_KmerScore"
"gene_A_3" "gene_A" "3" "0.0000" "92" "87" NA
"gene_A_4" "gene_A" "4" "0.0000" "92" "87" NA
"gene_A_5" "gene_A" "5" "0.0000" "92" "87" 0
"gene_A_6" "gene_A" "6" "0.0000" "93" "88" 0.0014
"gene_A_7" "gene_A" "7" "0.0000" "95" "89" 0.0027
"gene_A_8" "gene_A" "8" "0.0014" "95" "89" 0.004
Nanopolish results are stored in nanopolish-compore_flow directory. It contains two files per sample: raw eventalign output (gzipped) and another with the median raw current per position and transcript (sample_processed_perpos_median.tsv.gz). See example below:
contig position reference_kmer read_name median coverage
gene_A 0 AAATT 1 113.35 433
gene_A 1 AATTG 1 97.24 506
gene_A 2 ATTGA 1 70.35 2034
gene_A 3 TTGAA 1 102.03 416
gene_A 4 TGAAG 1 115.315 422
gene_A 5 GAAGA 1 104.25 471
Nanocompore results are stored in nanopolish-compore_flow directory. It contains one file per comparison (wt_1_vs_ko_1_nanocompore_results.tsv). Default output from Nanocompore (see Nanocompore’s repository for a more detailed explanation).
Encoding of modification information from m6A-aware basecalled data using modPhred
Once the data has been basecalled with our m6A modification-aware basecalling model, modification information data should be encoded for its later downstream analysis. This step is performed by modPhred, another software included in the mop_mod module.
To run this software, in the params.yaml file you should specify modphred: "YES" and run the code below:
cd mop_mod
nextflow run mop_mod.nf -params-file params.yaml -with-singularity -bg > yourlog.txt