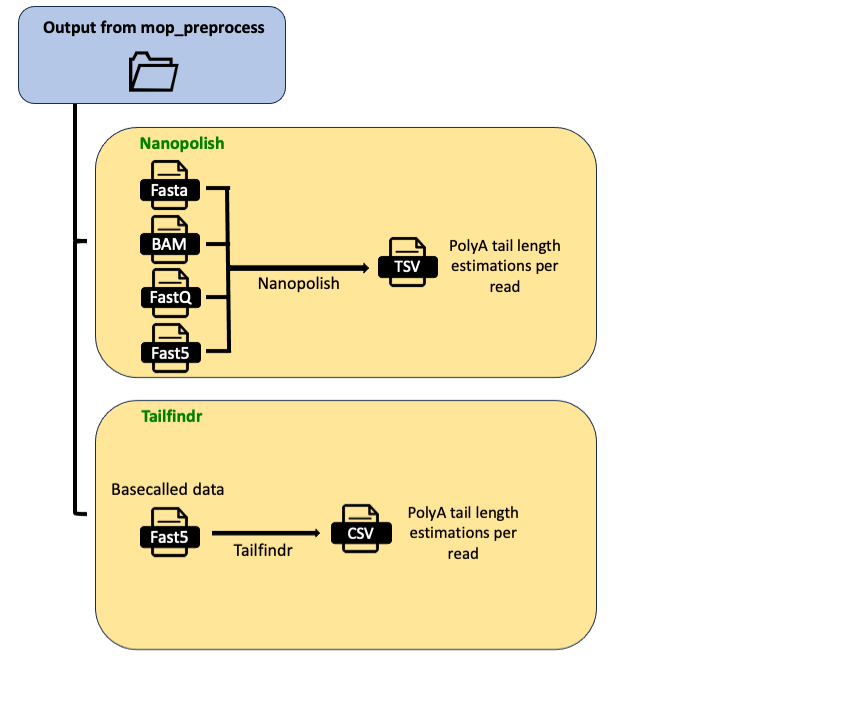
MOP_TAIL
This pipeline takes as input the output from MOP_PREPROCESS: basecalled fast5 reads, together with their respective fastq files, alignment and assignment read ID to gene/transcript. It outputs the estimation of poly(A) tail length at read level provided by Tailfindr, Nanopolish or both. Tailfinr can be run using three modes: standard, for Nano3P-seq protocol with R9 chemistry and Nano3P-seq protocol with R10 chemistry.
Input Parameters
The input parameters are stored in yaml files like the one represented here:
input_path: "${projectDir}/../mop_preprocess/outfolder/"
reference: "${projectDir}/../anno/yeast_rRNA_ref.fa.gz"
pars_tools: "${projectDir}/tools_opt.tsv"
output: "${projectDir}/outputPoly"
tailfindr: "YES"
# Different modes: standard, n3ps_r9 or n3ps_r10
tailfindr_mode: "standard"
nanopolish: "YES"
email: "yourname@yourdomain"
How to run the pipeline
Before launching the pipeline,user should:
Decide which containers to use - either docker or singularity [-with-docker / -with-singularity].
Fill in both params.config and tools_opt.tsv files.
To launch the pipeline, please use the following command:
nextflow run mop_tail.nf -params-file params.yaml -with-singularity > log.txt
You can run the pipeline in the background adding the nextflow parameter -bg:
nextflow run mop_tail.nf -params-file params.yaml -with-singularity -bg > log.txt
You can change the parameters either by changing params.config file or by feeding the parameters via command line:
nextflow run mop_tail.nf -params-file params.yaml -with-singularity -bg --output test2 > log.txt
You can specify a different working directory with temporary files:
nextflow run mop_tail.nf -params-file params.yaml -with-singularity -bg -w /path/working_directory > log.txt
Results
Several folders are created by the pipeline within the output directory specified by the output parameter:
NanoPolish: contains the output of nanopolish tool.
Tailfindr: contains the output of tailfindr tool.
PolyA_final: contains the txt files with the combined results (i.e. predicted polyA sizes). Here an example of a test:
"Read name" "Tailfindr" "Nanopolish" "Gene Name"
"013a5dde-9c52-4de1-83eb-db70fb2cd130" 52.16 49.39 "YKR072C"
"01119f62-ca68-458d-aa1f-cf8c8c04cd3b" 231.64 274.28 "YDR133C"
"0154ce9c-fe6b-4ebc-bbb1-517fdc524207" 24.05 24.24 "YFL044C"
"020cde28-970d-4710-90a5-977e4b4bbc46" 41.27 56.79 "YGL238W"
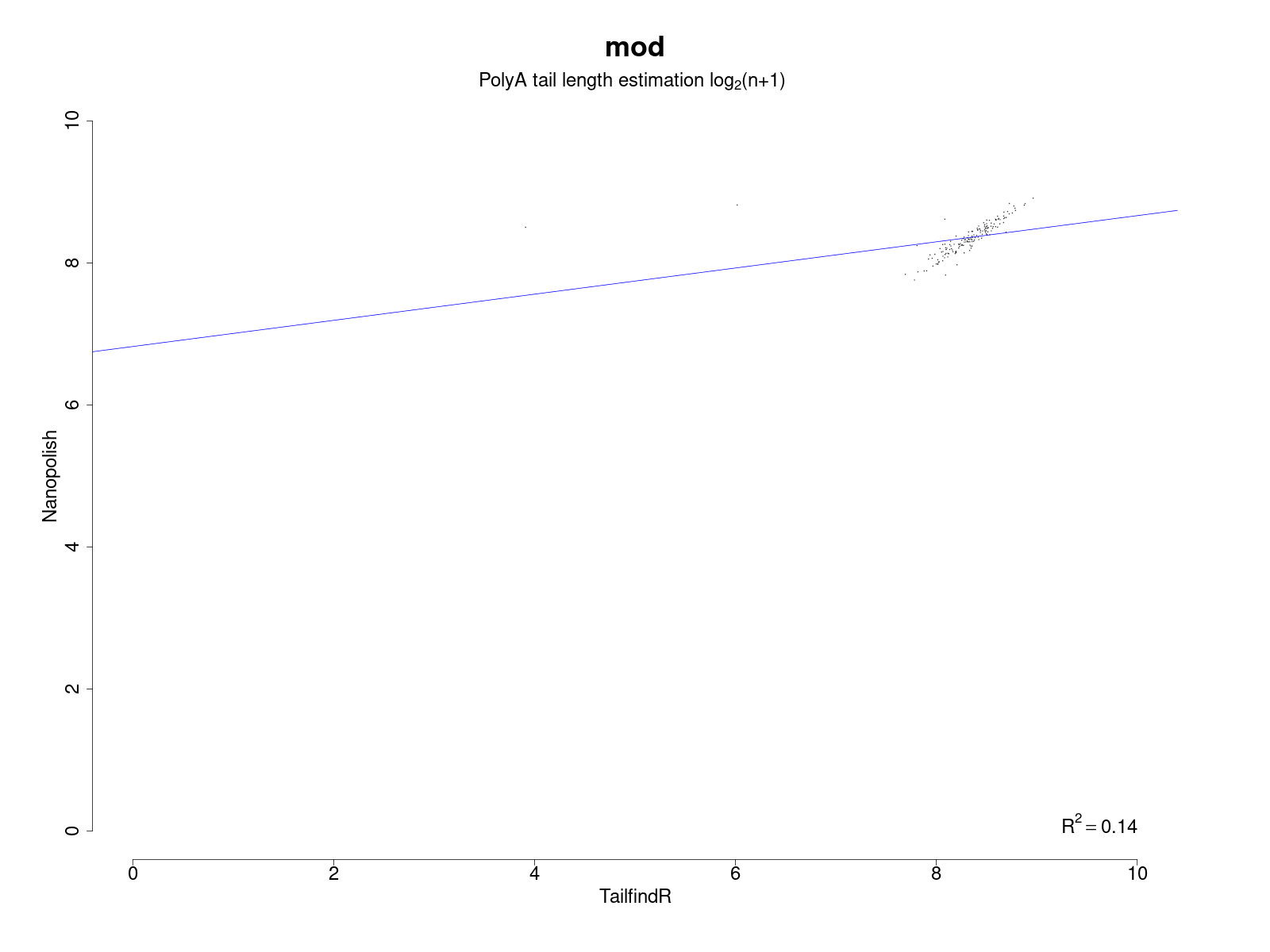
If both programs are run, an additional plot that shows the correlation of their results is generated.