

NanoPreprocess
This module takes as input the raw fast5 reads - single or multi - and produces a number of outputs (basecalled fast5, sequences in fastq format, aligned reads in BAM format etc). The pre-processing module performs base-calling, demultiplexing (optional), filtering, quality control, mapping to a genome / transcriptome reference, feature counting and it generates a final report of the performance and results of each of the steps performed. It automatically detects the kind of input fast5 file (single or multi sequence).
Workflow
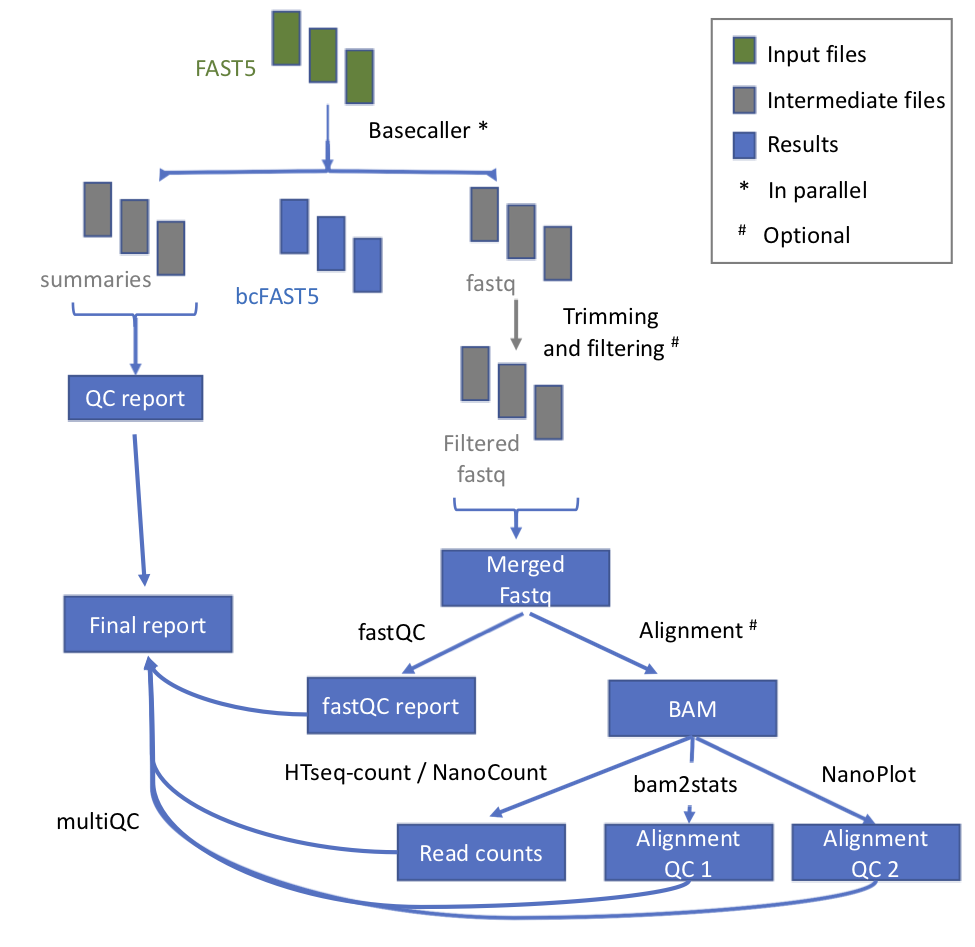
| Process name | Description |
|---|---|
| testInput | Detection of kind of fast5 (multi or single) |
| baseCalling | Basecalling with Albacore or Guppy (up to guppy 4.0) |
| demultiplexing | Demultiplexing (optional) |
| concatenateFastQFiles | This process concatenates the fastq files produces for each single basecalling |
| QC | Performed with MinIONQC |
| fastQC | Executed on fastq files |
| mapping | Mapping to genome / transcriptome with either minimap2, graphmap orgraphmap2 |
| counting | If mapping to the genome, it obtains counts per gene with htseq-count. Otherwise, if mapping to the transcriptome, transcript counts are generated with NanoCount. Reads are also assigned to a gene or to a transcript if they are uniquely mapping. A report file is also generated. |
| alnQC2 | QC of aligned reads with NanoPlot. The plots PercentIdentityvsAverageBaseQuality_kde, LengthvsQualityScatterPlot_dot, HistogramReadlength and Weighted_HistogramReadlength are then merged together in a single picture. |
| alnQC | QC of aligned reads with bam2stats. |
| cram_conversion | Generating cram file from alignment. |
| joinAlnQCs | Merging the QC files generated by the alnQC step. |
| joinCountQCs | Merging the report files generated by the counting step. |
| multiQC | Final report generation - enventually sent by mail to the user too. |
Input Parameters
| Parameter name | Description |
|---|---|
| fast5 files | Path to fast5 input files (single or multi-fast5 files). They should be inside a folder that will be used as sample name. [/Path/sample_name/*.fast5] |
| reference | File in fasta format. [Reference_file.fa] |
| ref_type | Specify if the reference is a genome or a transcriptome. [genome / transcriptome] |
| kit | Kit used in library prep - required for basecalling. |
| flowcell | Flowcell used in sequencing - required for basecalling. |
| annotation | Annotation file in GTF format. It is optional and needed only in case of mapping to the genome and when interested in gene counts. [Annotation_file.gtf] |
| seq_type | Sequence type. [RNA / DNA] |
| output | Output folder name. [/Path/to_output_folder] |
| granularity | indicates the number of input fast5 files analyzed in a single process. It is by default 4000 for single-sequence fast5 files and 1 for multi-sequence fast5 files. In case GPU option is turned on this value is not needed since every file will be analyzed sequentially. |
| basecaller | Algorithm to perform the basecalling. guppy or albacore are supported. [albacore / guppy] |
| basecaller_opt | Command line options for basecaller program. Check available options in respective basecaller repository. |
| GPU | Allow the pipeline to run with GPU. [OFF / ON] |
| demultiplexing | Demultiplexing algorithm to be used. [OFF / deeplexicon / guppy / guppy-readucks] |
| demultiplexing_opt | Command line options for the demultiplexing software. |
| demulti_fast5 | If performing demultiplexing, also generate demultiplexed multifast5 files. [OFF / ON] |
| filter | Program to filter fastq files. [nanofilt / OFF] |
| filter_opt | Command line options of the filtering program. |
| mapper | Mapping algorithm. [minimap2 / graphmap / graphmap2] |
| mapper_opt | Command line options of the mapping algorithm. |
| map_type | Spliced - recommended for genome mapping - or unspliced - recommended for transcriptome mapping. [spliced / unspliced] |
| counter | Generating gene counts (genome mapping) or transcript counts (transcriptome mapping). [YES / “”] |
| counter_opt | Command line options of the counter program: NanoCount for transcripts and Htseq-count for genes. |
| Users email for receving the final report when the pipeline is finished. [user_email] |
You can change them by editing the params.config file or using the command line - please, see next section.
How to run the pipeline
Before launching the pipeline, user should decide which containers to use - either docker or singularity [-with-docker / -with-singularity]. Then, to launch the pipeline, please use the following command:
nextflow run nanopreprocess.nf -with-singularity > log.txt
- Run the pipeline in the background:
nextflow run nanopreprocess.nf -with-singularity -bg > log.txt - Run the pipeline while changing params.config file:
nextflow run nanopreprocess.nf -with-singularity -bg --output test2 > log.txt - Specify a directory for the working directory (temporary files location):
nextflow run nanopreprocess.nf -with-singularity -bg -w /path/working_directory > log.txt - Run the pipeline with GPU - CRG GPU cluster users
nextflow run nanopreprocess.nf -with-singularity -bg -w /path/working_directory -profile cluster > log.txt - Run the pipeline with GPU - local GPU
nextflow run nanopreprocess.nf -with-singularity -bg -w /path/working_directory -profile local > log.txt
Troubleshooting
-
Checking what has gone wrong:
If there is an error, please see the log file (log.txt) for more details. Furthermore, if more information is needed, you can also find the working directory of the process in the file. Then, access that directory and check both the.command.logand.command.errfiles. -
Resume an execution:
Once the error has been solved or if you change a specific parameter, you can resume the execution with the Netxtlow parameter -resume (only one dash!). If there was an error, the pipeline will resume from the process that had the error and proceed with the rest. If a parameter was changed, only processes affected by this parameter will be re-run.
nextflow run nanopreprocess.nf -with-singularity -bg -resume > log_resumed.txt
To check whether the pipeline has been resumed properly, please check the log file. If previous correctly executed process are found as Cached, resume worked!
...
[warm up] executor > crg
[e8/2e64bd] Cached process > baseCalling (RNA081120181_1)
[b2/21f680] Cached process > QC (RNA081120181_1)
[c8/3f5d17] Cached process > mapping (RNA081120181_1)
...
IMPORTANT: To resume the execution, temporary files generated previously by the pipeline must be kept. Otherwise, pipeline will re-start from the beginning.
Results:
Several folders are created by the pipeline within the output directory specified by the output parameter and the input folder name is taken as sample name.
- fast5_files: Contains the basecalled multifast5 files. Each batch contains 4000 sequences.
- fastq_files: Contains one or, in case of demultiplexing, more fastq files.
- QC_files: Contains each single QC produced by the pipeline.
- alignment: Contains the bam file(s).
- cram_files: Contains the cram file(s).
- counts (OPTIONAL): Contains read counts per gene / transcript if counting was performed.
- assigned (OPTIONAL): Contains assignment of each read to a given gene / transcript if counting was performed.
- report: Contains the final multiqc report.
- variants (OPTIONAL): still experimental. It contains variant calling.
NanoPreprocessSimple
This is a light version of NanoPreprocess that does not perform the basecalling step. It allows to make the same analysis starting from basecalled reads in fastq format. You can also provide fast5 files if you need to demultiplex using DeepLexiCon.
This module will allow to run the pipeline on multiple input samples by using this syntax in the params.file:
fastq = "$baseDir/../../../org_data/**/*.fastq.gz"
In this way it will produces a number of output files with the same sample name indicated by the two asterisks.