

NanoTail
This module allows to estimates polyA sizes by using two different methods (nanopolish and talifindr). it reads directly the output produced by NanoPreprocess and in particular it needs the read counts / assignment.
Workflow
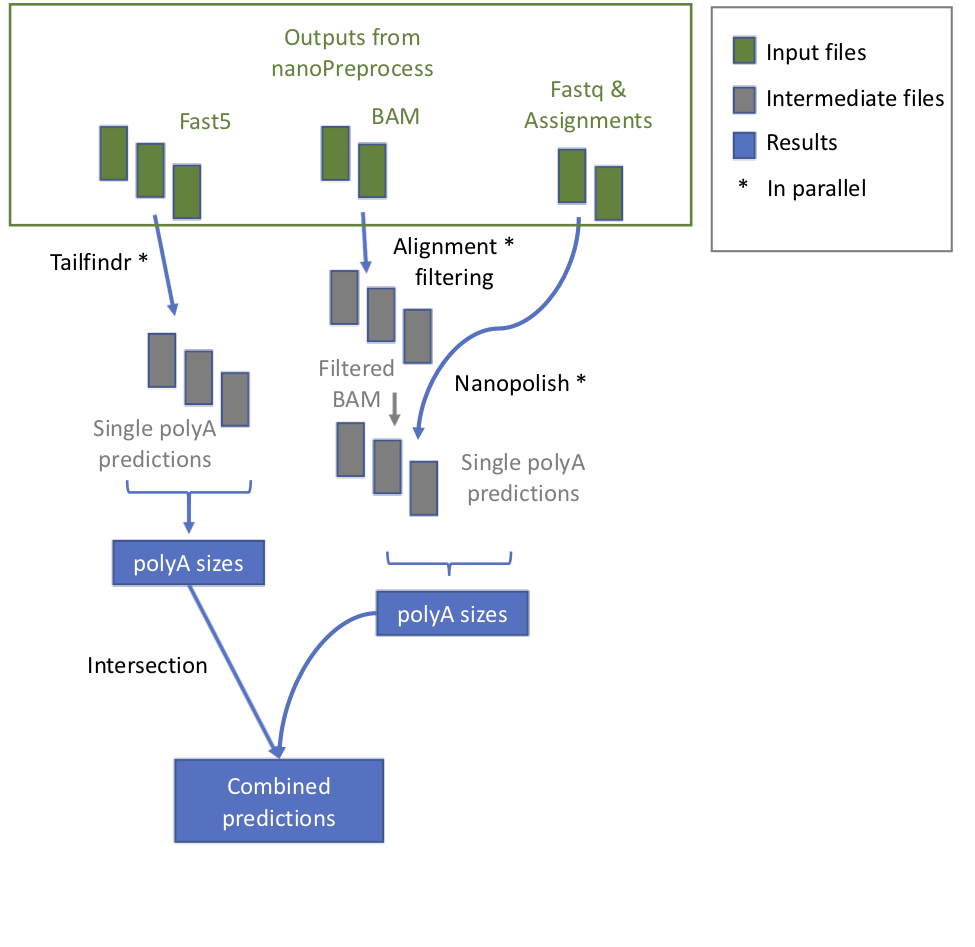
- check_reference It verifies whether the reference is zipped and eventually unzip it
- tailfindr it runs tailfindr tool in parallel.
- collect_tailfindr_results It collects the results of tailfindr.
- filter_bam Bam files are filtered with samtools to keep only mapped reads and remove secondary alignments
- tail_nanopolish It runs nanopolish in parallel.
- collect_nanopolish_results It collects the results of tail_nanopolish.
- join_results It merges the results from the two algorithms and make a plot of the correlation.
Input Parameters
- input_folders path to the folders produced by NanoPreprocessing step.
- nanopolish_opt options for the nanopolish program
- tailfindr_opt options for the tailfindr program
- reference reference genome / transcriptome
- output folder
Results
Three folders are created by the pipeline within the output folder:
- NanoPolish: contains the output of nanopolish tool.
- Tailfindr: contains the output of tailfindr tool.
- PolyA_final: contains the txt files with the combined results (i.e. predicted polyA sizes). Here an example of a test:
"Read name" "Tailfindr" "Nanopolish" "Gene Name"
"013a5dde-9c52-4de1-83eb-db70fb2cd130" 52.16 49.39 "YKR072C"
"01119f62-ca68-458d-aa1f-cf8c8c04cd3b" 231.64 274.28 "YDR133C"
"0154ce9c-fe6b-4ebc-bbb1-517fdc524207" 24.05 24.24 "YFL044C"
"020cde28-970d-4710-90a5-977e4b4bbc46" 41.27 56.79 "YGL238W"
A plot is also produced for showing the correlation between the two methods.