

Pre-processing of sequencing reads
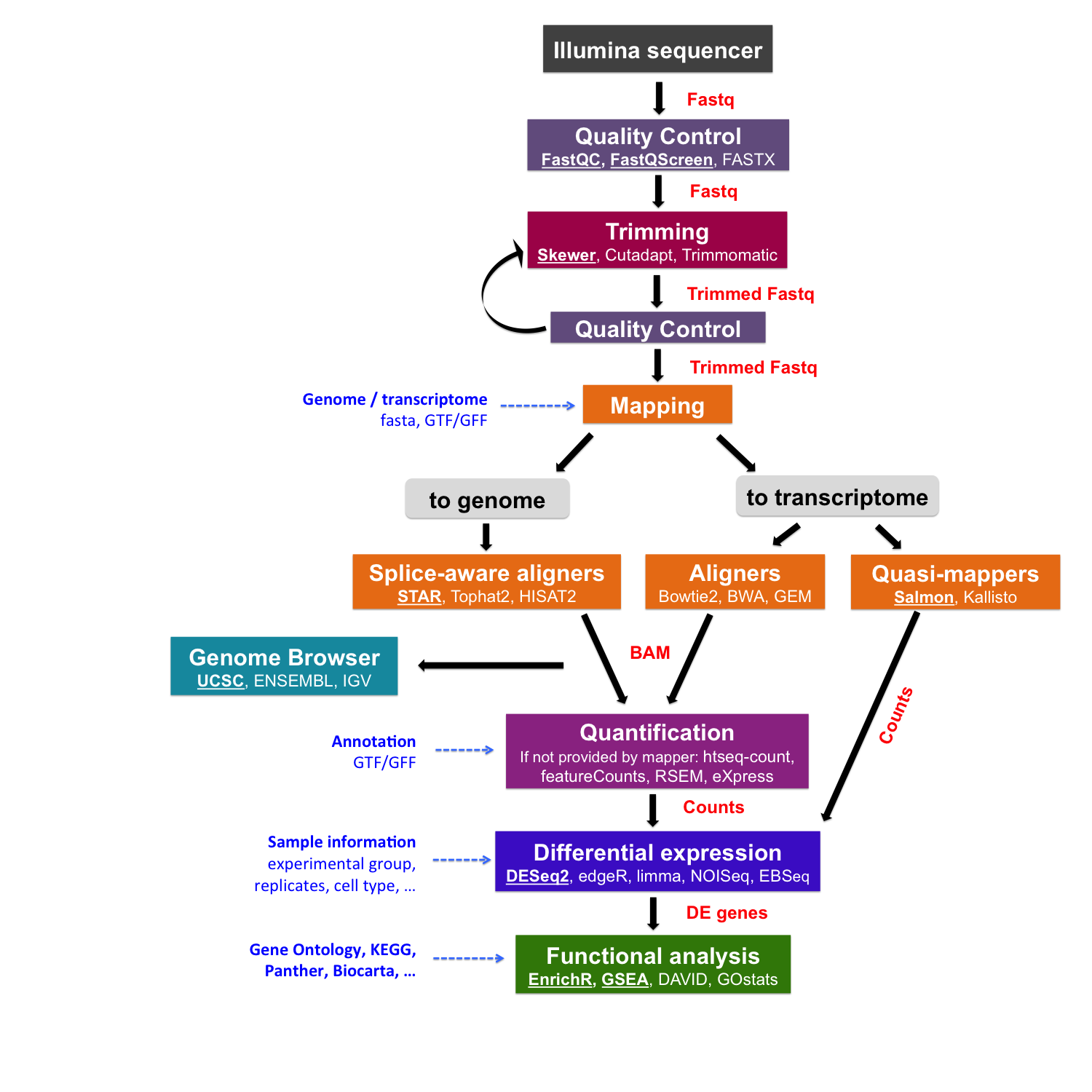
Once sequencing reads are obtained from the sequencing machine, they need to be pre-processed for further analysis. This step includes the quality control of initial reads and read trimming that includes removing adapter sequences, filtering out low quality reads and trimming reads off low quality base pairs.
QC of sequencing reads
To assess the quality of sequencing data, we will use the programs FastQC and Fastq Screen.
FastQC calculates statistics about the composition and quality of raw sequences, while Fastq Screen looks for possible contaminations.
mkdir QC
$RUN fastqc resources/A549_25_3chr10_*.fastq.gz -o ./QC/
Started analysis of A549_25_3chr10_1.fastq.gz
Approx 5% complete for A549_25_3chr10_1.fastq.gz
Approx 10% complete for A549_25_3chr10_1.fastq.gz
Approx 15% complete for A549_25_3chr10_1.fastq.gz
Approx 20% complete for A549_25_3chr10_1.fastq.gz
...
Approx 85% complete for A549_25_3chr10_2.fastq.gz
Approx 90% complete for A549_25_3chr10_2.fastq.gz
Approx 95% complete for A549_25_3chr10_2.fastq.gz
Analysis complete for A549_25_3chr10_2.fastq.gz
We can display the results with a browser; e.g., Firefox, for each file individually or all files with one command:
firefox QC/A549_25_3chr10_1_fastqc.html
firefox QC/*.html
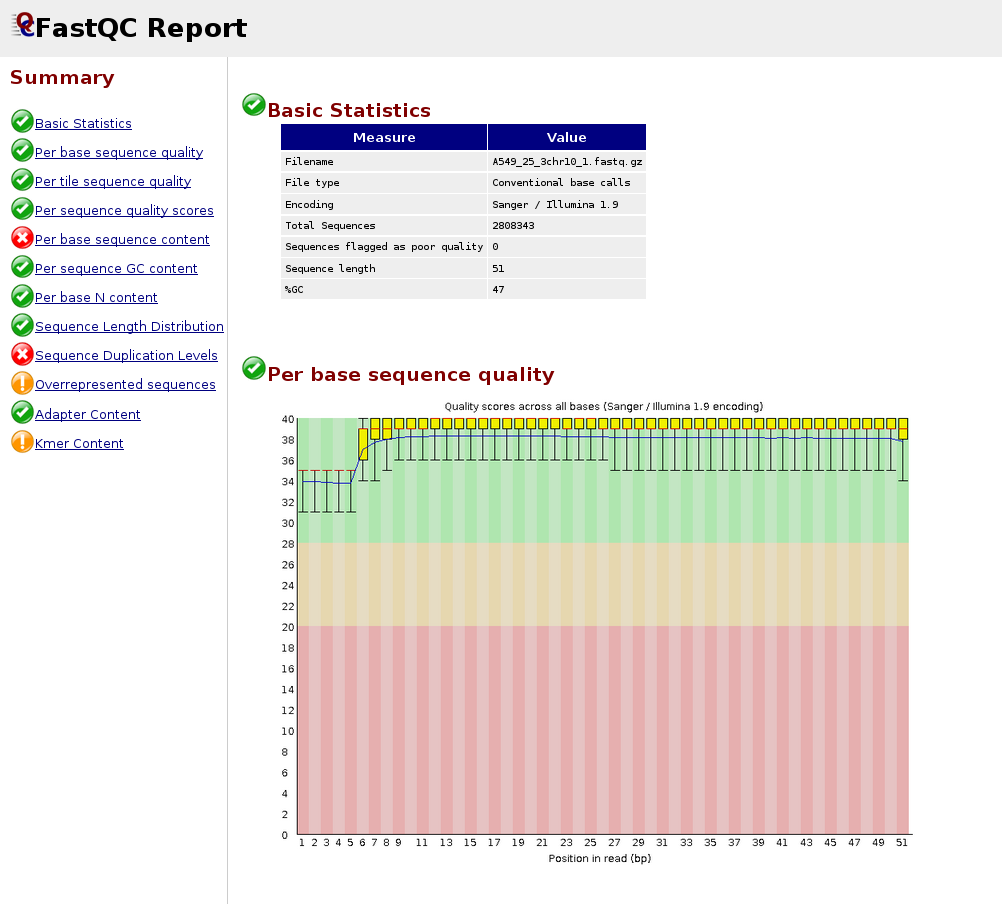
Below is an example of a poor quality dataset. As you can see, the average quality drops dramatically towards the 3’-end.
FastQ Screen requires a number of databases to be installed. Note, the program is included in the Docker and Singularity images but the databases are not. A number of databases prepared by the authors of the program can be downloaded by using the following command (DO NOT LAUNCH IT IN THE CLASS because it will take a while!!! But you will need to do it when completing the Final project):
$RUN fastq_screen --get_genomes
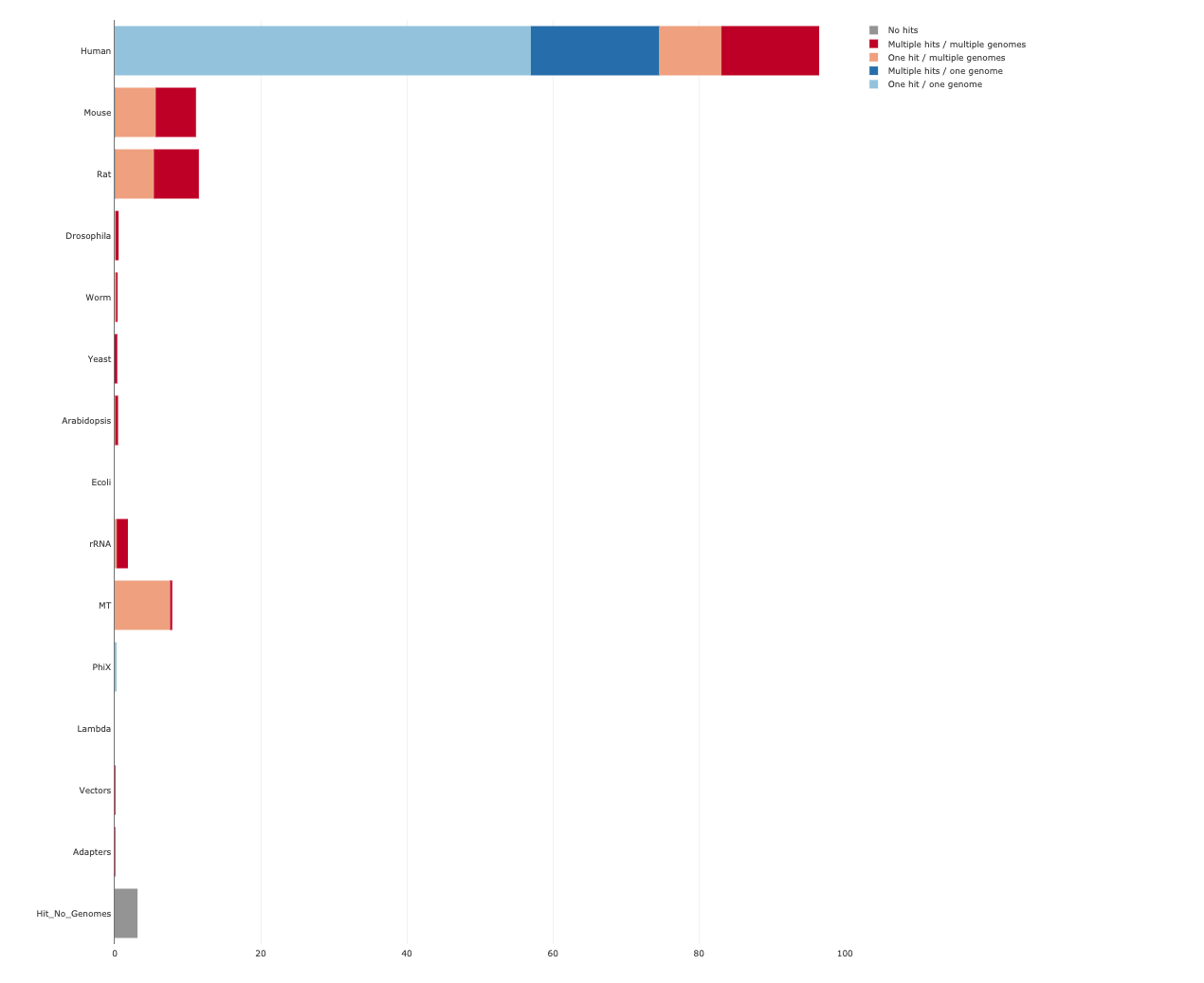
This will download Bowtie indexes for 11 genomes (arabidopsis, drosophila, E. coli, human, lambda, mouse, mitochondria, phiX, rat, worm and yeast) and 3 collection of sequences (adapters, vectors, rRNA). The files will be downloaded in the FastQ_Screen_Genomes folder. The file fastq_screen.conf will be also downloaded in this folder. To use the tool, you will have to modify the fastq_screen.conf by providing the full path to the Bowtie2 executable (/usr/local/bin/bowtie2 if you use our singularity image) and full paths to the downloaded folders with genome index files. Here we show where to change the executables:
# This is a configuration file for fastq_screen
###########
## Bowtie #
###########
## If the bowtie binary is not in your PATH then you can
## set this value to tell the program where to find it.
## Uncomment the line below and set the appropriate location
##
#BOWTIE /usr/local/bin/bowtie/bowtie
BOWTIE2 /usr/local/bin/bowtie2
...
FastQ Screen runs check on a random subset of 100,000 reads (that can be changed using option –subset).
To execute FastQ Screen:
$RUN fastq_screen --conf FastQ_Screen_Genomes/fastq_screen.conf \
resources/A549_0_1chr10_1.fastq.gz \
--outdir ./QC/A549_0_1
Using fastq_screen v0.13.0
Reading configuration from 'fastq_screen.conf'
Aligner (--aligner) not specified, but Bowtie2 path and index files found: mapping with Bowtie2
Adding database Human
Adding database Mouse
Adding database Rat
Adding database Drosophila
Adding database Worm
Adding database Yeast
Adding database Arabidopsis
Adding database Ecoli
Adding database rRNA
Adding database MT
Adding database PhiX
Adding database Lambda
Adding database Vectors
Adding database Adapters
Using 7 threads for searches
Option --subset set to 100000 reads
Processing A549_0_1chr10_1.fastq.gz
Counting sequences in A549_0_1chr10_1.fastq.gz
Making reduced sequence file with ratio 711:1
...
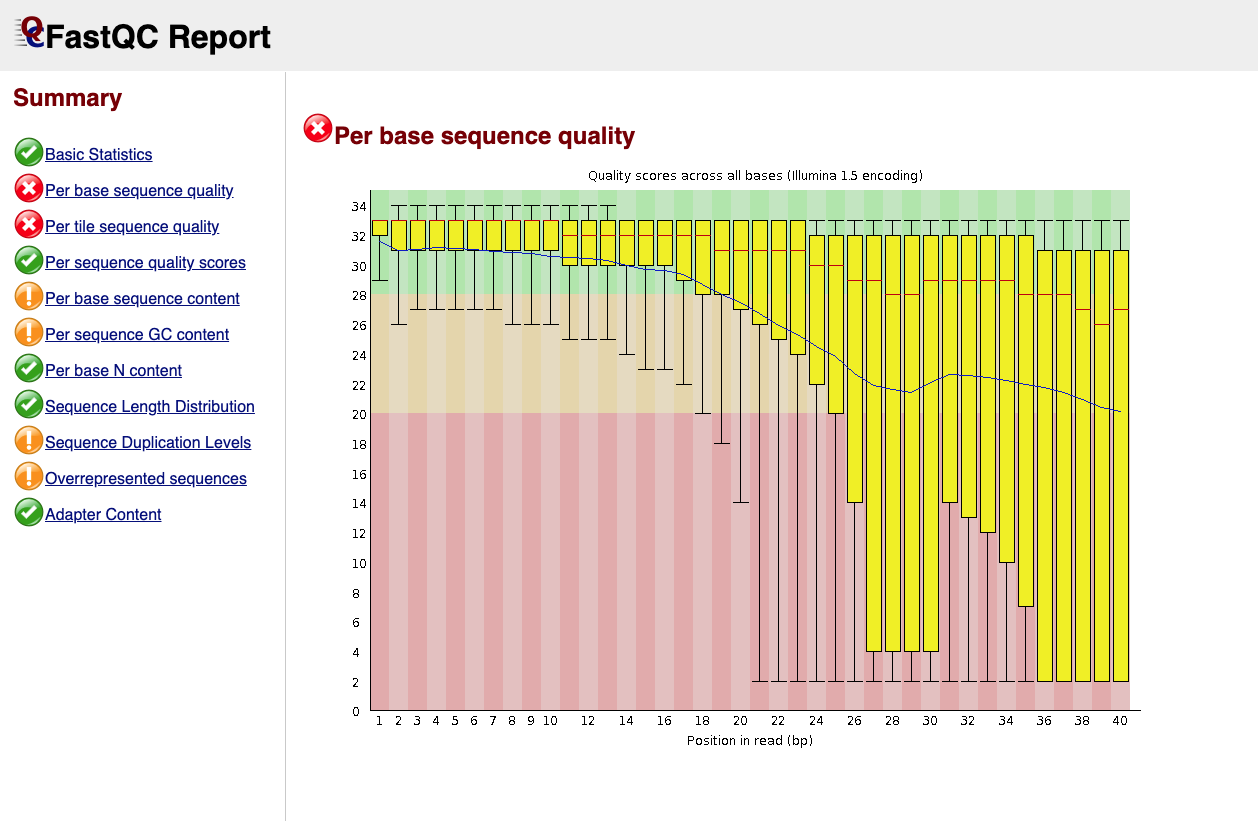
Below is an example of the FastQ Screen results for A549_0_1_1.fastq.gz which we prepared.
wget https://biocorecrg.github.io/RNAseq_course_2019/precomp_res/A549_0_1_fastq_screen.tar.gz
tar -zvxf A549_0_1_fastq_screen.tar.gz
A549_0_1_fastq_screen/
A549_0_1_fastq_screen/A549_0_1chr10_1_screen.html
A549_0_1_fastq_screen/A549_0_1chr10_1_screen.txt
A549_0_1_fastq_screen/A549_0_1chr10_1_screen.png
mv A549_0_1_fastq_screen QC
firefox QC/A549_0_1_fastq_screen/A549_0_1chr10_1_screen.html
Initial processing of sequencing reads
Before mapping reads to the genome/transcriptome or performing a de novo assembly, the reads has to be pre-processed, if needed, as follows:
- Demultiplex by index or barcode (it is usually done in the sequencing facility)
- Remove adapter sequences
- Trim reads by quality
- Discard reads by quality/ambiguity
- Filter reads by k-mer coverage (recommended for the de novo assembly)
- Normalize k-mer coverage (recommended for the de novo assembly)
As shown before, both the presence of low quality reads and adapters are reported in the fastqc output.
Adapters are usually expected in small RNA-Seq because the molecules are typically shorter than the reads, and that makes an adapter to be present at 3’-end. Let’s run FastQC on a fastq file for small RNA-Seq.
$RUN fastqc resources/subsample_to_trim.fq.gz -o ./QC
Started analysis of subsample_to_trim.fq.gz
Approx 5% complete for subsample_to_trim.fq.gz
Approx 10% complete for subsample_to_trim.fq.gz
Approx 15% complete for subsample_to_trim.fq.gz
Approx 20% complete for subsample_to_trim.fq.gz
Approx 25% complete for subsample_to_trim.fq.gz
...
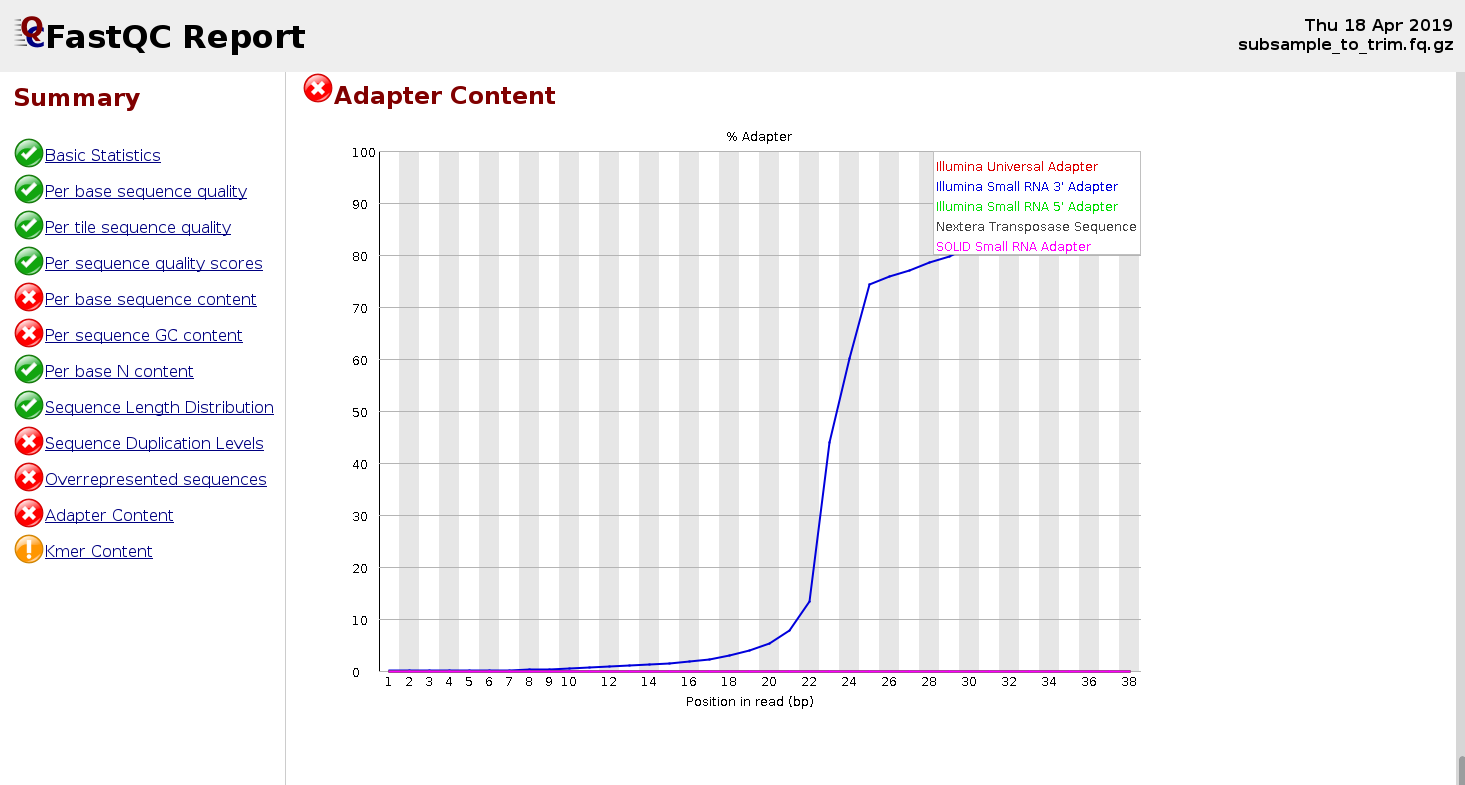
EXERCISE
Once you got the FastQC report (above), how to figure out the sequence(s) of the adapter(s) that needs to be trimmed?
There are many tools for trimming reads and removing adapters, such as Trim Galore!, Trimmomatic, Cutadapt, skewer, AlienTrimmer, BBDuk, and the most recent SOAPnuke and fastp.
Let’s use skewer to trim the Illumina small RNA 3’ adapter.
$RUN skewer resources/subsample_to_trim.fq.gz -x TGGAATTCTCGGGTGCCAAGG -o QC/subsample_to_trim
.--. .-.
: .--': :.-.
`. `. : `'.' .--. .-..-..-. .--. .--.
_`, :: . `.' '_.': `; `; :' '_.': ..'
`.__.':_;:_;`.__.'`.__.__.'`.__.':_;
skewer v0.2.2 [April 4, 2016]
Parameters used:
-- 3' end adapter sequence (-x): TGGAATTCTCGGGTGCCAAGG
-- maximum error ratio allowed (-r): 0.100
-- maximum indel error ratio allowed (-d): 0.030
-- minimum read length allowed after trimming (-l): 18
-- file format (-f): Sanger/Illumina 1.8+ FASTQ (auto detected)
-- minimum overlap length for adapter detection (-k): 3
Thu Apr 18 17:51:18 2019 >> started
|=================================================>| (100.00%)
Thu Apr 18 17:51:25 2019 >> done (6.789s)
1000000 reads processed; of these:
30171 ( 3.02%) short reads filtered out after trimming by size control
2220 ( 0.22%) empty reads filtered out after trimming by size control
967609 (96.76%) reads available; of these:
958360 (99.04%) trimmed reads available after processing
9249 ( 0.96%) untrimmed reads available after processing
log has been saved to "QC/subsample_to_trim-trimmed.log".
We can look at the read distribution after the trimming of the adapter by inspecting the log-file or re-launching FastQC.
$RUN fastqc QC/subsample_to_trim-trimmed.fastq -o QC
Started analysis of subsample_to_trim-trimmed.fastq
Approx 5% complete for subsample_to_trim-trimmed.fastq
Approx 10% complete for subsample_to_trim-trimmed.fastq
Approx 15% complete for subsample_to_trim-trimmed.fastq
Approx 20% complete for subsample_to_trim-trimmed.fastq
Approx 25% complete for subsample_to_trim-trimmed.fastq
Approx 30% complete for subsample_to_trim-trimmed.fastq
...
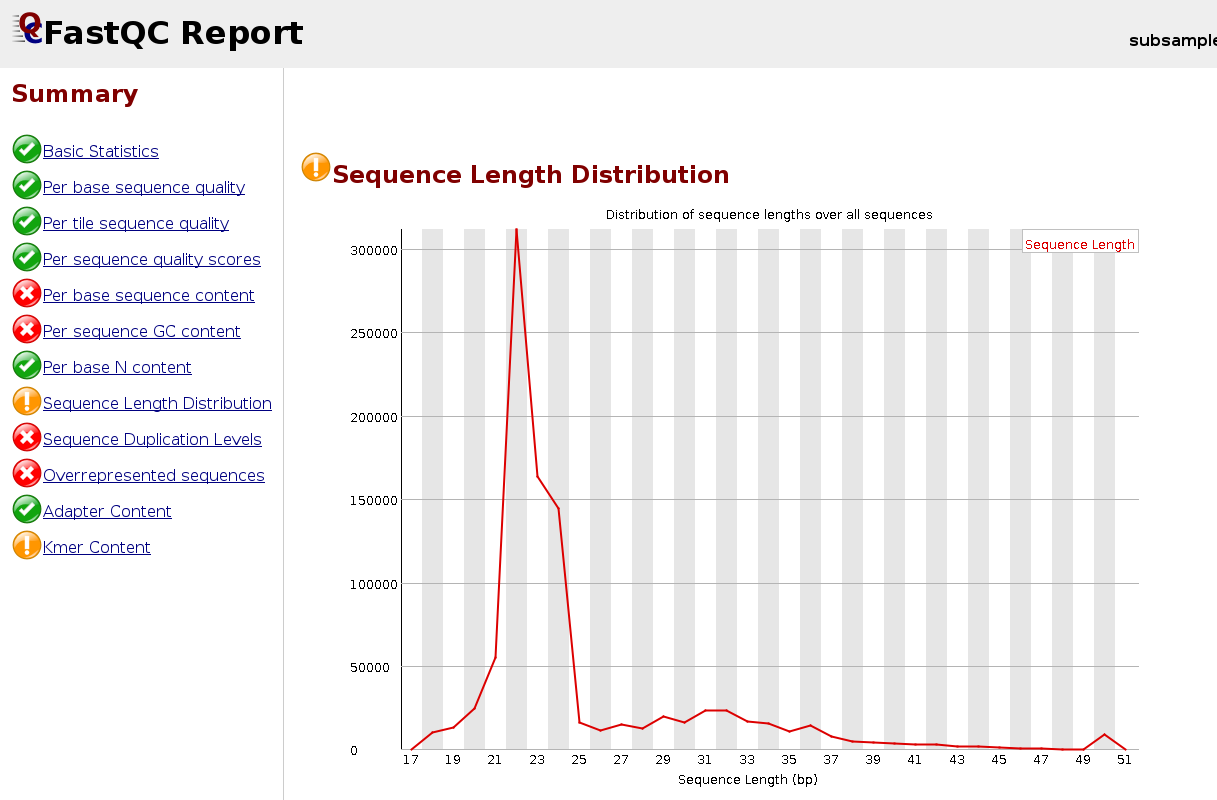
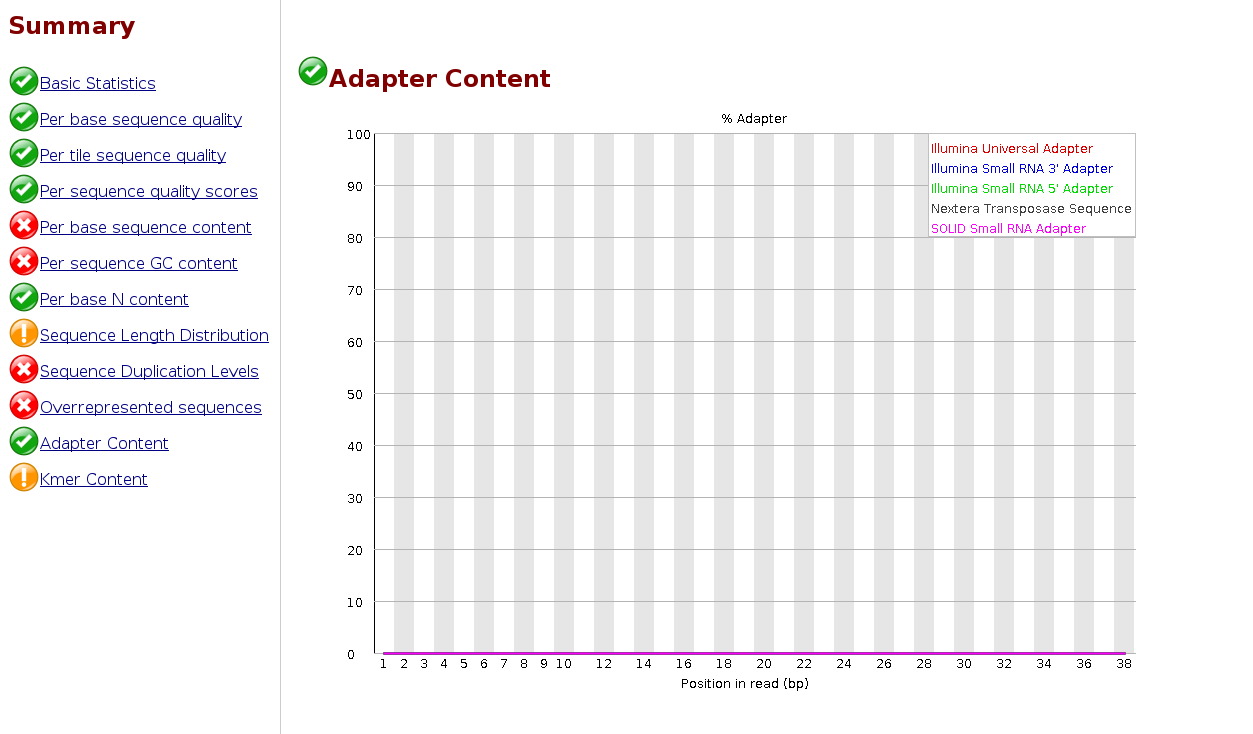
EXERCISE
Let’s explore the tool skewer in more detail, using “skewer –help” command.
- Which parameter indicates the minimum read length allowed after trimming? And what is its default value?
- Which parameter indicates the threshold on the average read quality to be filtered out?
- Using skewer filter out reads in “subsample_to_trim.fq-trimmed.fastq” that have average quality below 30 and trim them on 3’-end until the base quality is reached 30. How many reads were filtered out and how many remained?