

Mapping using Salmon
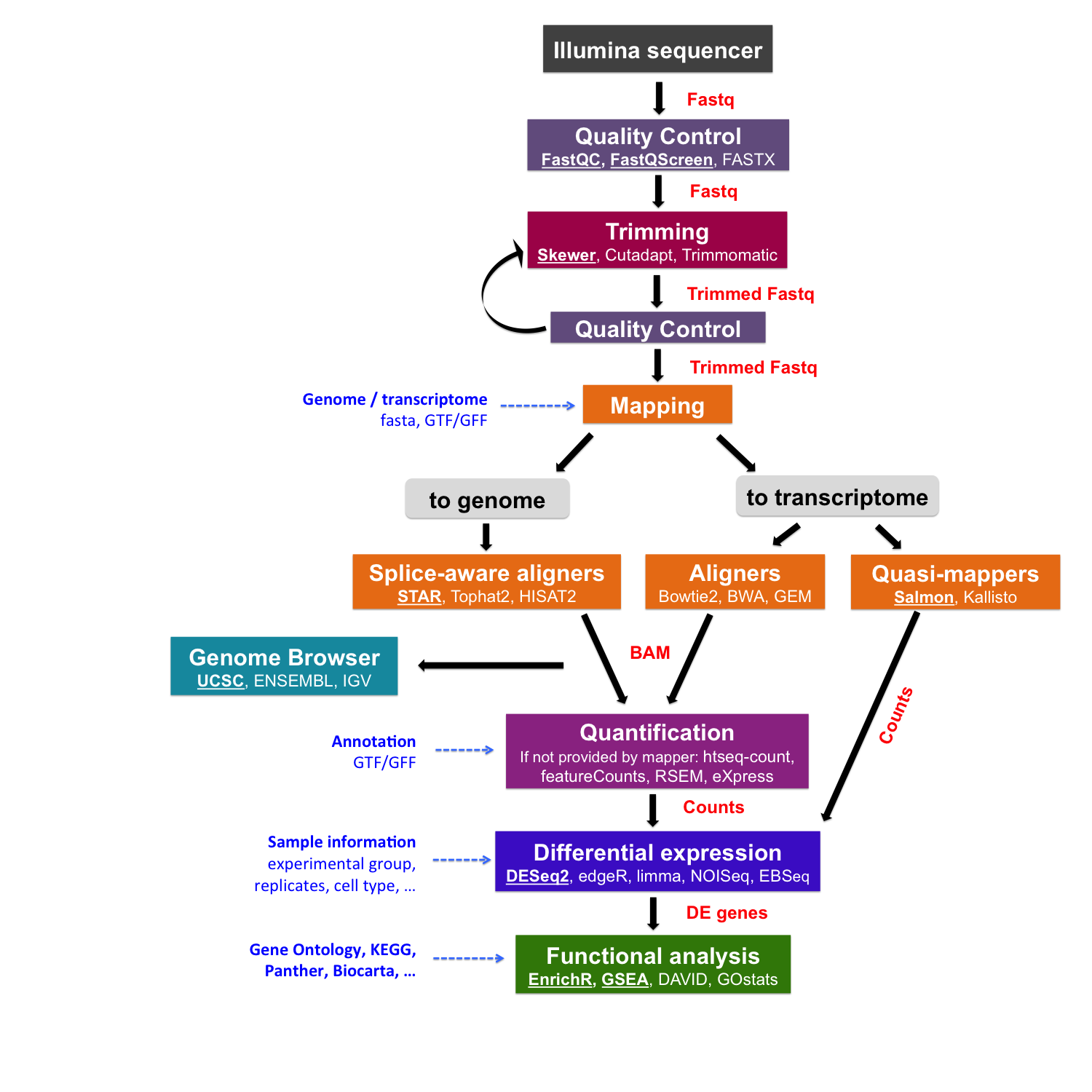
Salmon is a tool for quantifying the expression of transcripts using RNA-seq data. It is a quasi-mapper as it doesn’t produce the read alignments (and doesn’t output BAM/SAM files). Salmon “quasi-maps” reads to the transcriptome rather than to the genome as STAR does. Salmon can also make use of pre-computed alignments (in the form of a SAM/BAM file) to the transcripts rather than the raw reads in the FASTQ format.
Building the Salmon index
To make an index for Salmon, we need transcript sequences in the FASTA format. Salmon does not need any decompression of the input, so we can index by using this command:
$RUN salmon index -t annotations/gencode.v29.transcripts.fa.gz \
-i indexes/transcripts --gencode
Version Info: This is the most recent version of salmon.
index ["transcripts"] did not previously exist . . . creating it
[2019-04-30 18:12:59.272] [jLog] [info] building index
[2019-04-30 18:12:59.275] [jointLog] [info] [Step 1 of 4] : counting k-mers
[....]
[2019-04-30 18:18:07.251] [jLog] [info] done building index
| We add also the parameter –gencode since our data come from Gencode version 29 and their header contains several identifiers separated by the character ** | **. This parameter allows the program to parse the header and keep only the transcript identifier. |
Quantifying transcript expression
To quantify reads with Salmon, we need to specify the type of the sequencing library, aka Fragment Library Types in Salmon, using three letters:
The first:
| Symbol | Meaning | Reads |
|---|---|---|
| I | inward | -> … <- |
| O | outward | <- … -> |
| M | matching | > … -> |
The second:
| Symbol | Meaning |
|---|---|
| S | stranded |
| U | unstranded |
The third:
| Symbol | Meaning |
|---|---|
| F | read 1 (or single-end read) comes from the forward strand |
| R | read 1 (or single-end read) comes from the reverse strand |
From the STAR output for read counts we already know that for the analyzed experiment the ISR (Inward, Stranded and Reverse) library was used. If we want to assign the reads to the genes (option -g) in addition to transcripts we have to provide a GTF file corresponding to the transcript version which was used to build the Salmon index. We can download the whole GTF file here:
cd annotations
wget wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_29/gencode.v29.annotation.gtf.gz
gunzip gencode.v29.annotation.gtf.gz
cd ../
Then we can proceed with the mapping.
$RUN salmon quant -i indexes/transcripts -l ISR \
-1 resources/A549_0_1chr10_1.fastq.gz \
-2 resources/A549_0_1chr10_2.fastq.gz \
-o alignments/salmon_A549_0_1 \
-g annotations/gencode.v29.annotation.gtf \
--seqBias \
--validateMappings \
-p 4
Version Info: This is the most recent version of salmon.
### salmon (mapping-based) v0.13.1
### [ program ] => salmon
### [ command ] => quant
....
We can check the results inside the folder “alignments”.
ls alignments/salmon_A549_0_1/
aux_info cmd_info.json lib_format_counts.json libParams logs quant.genes.sf quant.sf
For explanation of all output files, see the Salmon documentation. The most interesting to us in this course is the file quant.genes.sf, that is a tab-separated file containing the read counts for genes:
| Column | Meaning |
|---|---|
| Name | Gene name |
| Length | Gene length |
| EffectiveLength | Effective length after considering biases |
| TPM | Transcripts Per Million |
| NumReads | Estimated number of reads considering both univocally and multimapping reads |
head -n 5 alignments/salmon_A549_0_1/quant.genes.sf
Name Length EffectiveLength TPM NumReads
ENSG00000285803.1 1152 1116.21 7.96961 15.764
ENSG00000285712.1 1590 1545.58 2.19064 6
ENSG00000285824.1 1120 860.855 6.91601 10.551
ENSG00000285884.1 790 515.683 3.28285 3
...
There is also similar formatted file that provides read counts for transcript:
head -n 5 alignments/salmon_A549_0_1/quant.sf
Name Length EffectiveLength TPM NumReads
ENST00000016171.5 2356 1970.742 659.861626 2304.468
ENST00000020673.5 4183 5925.497 0.000000 0.000
ENST00000173785.4 925 868.802 0.000000 0.000
ENST00000181796.6 3785 3216.057 0.000000 0.000
...
We will use information on read counts for genes from quant.genes.sf files for the differential expression (DE) analysis.