

What you will learn today
- How function is formally defined and annotated.
- What is Gene Ontology.
- Functional domain vs. structural domain.
- How function is transferred to homologous proteins.
- Blast - an algorithm for sequence alignment and search. How to use it and understand the output.
- How to infer the protein function using sequence homology.
Protein function
- Major classes of biochemical functions:
- Binding, or specific recognition of other molecules (proteins, peptides, DNA, RNA, lipids, small molecules, metals) – TBP, myoglobin, MHC
- Catalysis (protein enzymes) – DNA polymerase, HIV protease
- Conformational switches in response to changes in pH or ligand binding – GTPase Ras, kinases
- Structural proteins – silk, F-actin
-
Function defined through cellular localization: nuclear receptors, membrane proteins, cytoskeletal proteins, telomerase, etc.
- Function defined through participating in a biological process: signaling, transport, development, etc.
Protein function is context-dependent
-
Cellular localization: e.g., metal-binding proteins folding in different compartments can bind different ions (PMID: 18948958)
-
Post-translational modifications may affect protein stability, structure, binding, and localization; they can be irreversible as ubiquitination and limited proteolysis or reversible (act as conformational switches) as glycosylation, methylation, phosphorylation and N-acetylation.
-
Binding of other ligands:
- Cofactors such as the heme group which defines the function of hemoglobin and cytochrome c;
- Zinc-finger-containing proteins cannot bind DNA without presence of an ions;
- Co-activators allosterically change the conformation from disordered to ordered in the Leucine-zipper DNA-binding domain of transcription factors.
Homology: Orthology, analogy, paralogy
- Ortholog: Same ancestor origin. Speciation event
- Analog: Not the same origin (despite having the same structure and/or function)
- Paralog: Same former ancestor origin, but duplication event also involved
| Gene phylogeny of histone H1 |
|---|
 |
| https://upload.wikimedia.org/wikipedia/commons/4/4b/Ortholog_paralog_analog_examples.svg |
Protein functional domain vs. structural domain
Protein structural domain is a compact unit of a 3D protein structure folded (presumably) independently in solution; it does not necessarily carry specific function.
Protein functional domain (described in such databases as Pfam, PRINTS, NCBI CDD, COGs, etc.) is evolutionary conserved protein region; not necessarily coincides with structural domain. It is an evolutionary recurrent and reused functional unit inferred by means of sequence or function similarity (in the absence of structural information) or experimentally identified whittling down a protein to its smallest functional fragment. Less vigorously defined than structural domain, may consist of one or more structural domains.
Approaches at predicting protein function
- Sequence-based approach:
- Homology-based transfer: BLAST, etc.
- Sequence motifs (aka pattern-based methods); e.g., to predict subcellular location based on the short signal peptide.
- Clustering of orthologs
- Machine-learning approaches, or classifiers
- “Guilt-by-association” approach using genomic (experimental and predicted) data:
- Phylogenomic profiling (co-evolution)
- Expression data
- Protein-protein interactions
- Protein-DNA interactions
- Phenotype data (knockouts, knockdowns)
- Post-translational modifications
- Protein subcellular localization data
- Structure-smilarity based approaches
Functional terms
Synonyms: keywords, glossaries, vocabularies, etc.
- UniProt controlled vocabulary
- UniProt keywords
- Enzyme nomenclature Database (EC)
- etc.
- COG
- KEGG Orthology
- Online Mendelian Inheritance in Man
- EC number (for enzymes)
Example: https://uniprot.org/uniprot/P99999 Which UniProt keywords describe this protein?
Ontologies
- Ontology: a more complex case of a controlled vocabulary: formal naming and definition of the categories, properties and relations between the concepts, data and entities that substantiate one, many or all domains of discourse. (Ref)
Gene Ontology
Consists of 3 ontologies:
- Molecular function
- Biological process
- Cellular location
Terms are linked by different kind of relationships: is a, part of, regulates, etc.
Example: https://uniprot.org/uniprot/P99999 Let’s explore GO Biological Process term “Mitochondrial electron transport, cytochrome c to oxygen”
EXERCISE 1:
- Q.1. How many proteins in UniProt are annoted with this GO term?
- Q.2. How many of them are human proteins?
- Q.3. How many human proteins were assigned with this GO term based on the evidence “sequence similarity evidence used in manual assertion”?
Other ontologies
Moonlighting proteins
- Databases: MoonProt && MultiTaskDB
- Example: P99999 - Cytochrome c
- It transfers electrons between Complexes III (Coenzyme Q - Cyt C reductase)
- Controlling apoptosis
- Example: P99999 - Cytochrome c
Protein function can be inferred by means of sequence homology, with the exceptions:
- Moonlighting (multifunctional) proteins or proteins carrying different functions in different cellular compartments.
- Paralogs (homologues sequences in the same species) – they are more likely to diverge functionally than orthologs.
- Multi-domain proteins: the functional role of a domain is not explicit or a homolog can matche to the other domain.
- Wrong annotation in the first place!
Yet,
- One residue change can disrupt a functional site and thus the protein function.
- Highly homologous sequences can have different functions: e.g., melanine deaminase and atrazine chlorohydrolase.
EXERCISE 2:
- Q.1. What is the sequence similarity of melanine deaminase and atrazine chlorohydrolase? Search UniProt for Atrazine chlorohydrolase. Select ATZA_PSESD and TRIA_ACIAI and click “Align”.
- Q.2. While Blast is running, explore similarities and differences in function of these two proteins.
Protein and DNA sequences alignment
- To search sequences in the databases.
- To annotate (i.e., assign function to) genes, proteins, genomes by means of transferring curated annotation of homologous genes/proteins. For example, active sites of enzymes, binding sites of protein receptors, cis-regulatory elements in DNA are evolutionary conserved at the sequence level.
Pairwise sequence alignment methods
-
Local alignment searches for the most similar regions in the two sequences. It can produce more than one alignment. It is best used for sequences that share some degree of similarity or of different lengths. Best suited for finding conserved elements.
-
Global alignment compares two sequences along the entire length. It is therefore best for highly similar sequences of approximately the same length.
Local alignment: nucleotide BLAST — USE Firefox or Chrome browser!
At NCBI BLAST website, select Nucleotide BLAST.
Further, check the box Align two or more sequences and check radio-button Program selection –> Somewhat similar sequences (blastn), and check the box below Show results in a new window.
Now let’s align these two nucleotide sequences.
tccCAGATATGTCAGGGGACACGAGcatgcagagac
aattgccgccgtcgttttcagCAGTTATGTCAGGGGACACGAGatc
We can see that the positions 4-25 of the first sequence are aligned with the positions 22-43 of the second sequence with 21 out of 22 aligned nucleotides being identical (identity 95%), with zero gaps.
The Blast score is equal 39. Let’s go back to the input page to see how the score is calculated. Expand Algorithm parameters.
You can see that the default scoring parameters are +2 for match and -3 for mismatch. As we have 21 matches and 1 mismatch the score is equal
39 = 2*21 - 3*1.
Now let’s introduce a gap in the second sequence (we remove A before GGGG).
tccCAGATATGTCAGGGGACACGAGcatgcagagac
aattgccgccgtcgttttcagCAGTTATGTCGGGGACACGAGatc
The score became 30 = 2*20 - 3*1 - 5*1 - 2*1, where -5 is a score for a gap opening and -2 for the gap of length 1.
And the sequence identity becomes 91% (20/22 nucleotides are perfectly aligned).
Global alignment
At NCBI BLAST website, select Global Align and paste the above two sequences. You can see that the algorithm attempts to align two sequences along their entire length, thus giving the low sequence identity and many gaps.
Local alignment: protein BLAST — USE Firefox or Chrome browser!
At NCBI BLAST website, select Protein BLAST. Further, check the box Align two or more sequences and check also the box below Show results in a new window.
Now let’s align the human and mouse major prion proteins typing their Accession numbers.
BAG32277
NP_001265185
The result:
Score = 381 bits(979) E-value = 4e-140 Identities = 226/255(89%) Gaps = 3/255(1%)
Now, let’s explore the default parameters of the method, clicking on the input page Algorithm parameters. You can see that except for the gap cost parameters all other parameters are different from those that Nucleotide BLAST uses.
Let’s first explore masking low-compexity regions. Check the box Low complexity regions.
Low-complexity region means a region of compositional bias; that is, a sequence composed of:
- homopolymeric runs (e.g., AAAAA),
- short-period repeats (e.g., SPSPSPSP), or
-
over-represention of several residues.
- NOTE: This filtering can eliminate statistically significant but biologically uninteresting reports from the blast output (e.g., hits against common acidic-, basic- or proline-rich regions), leaving the more biologically interesting regions of the query sequence available for specific matching against database sequences.*
The new result:
Score = 256 bits(655) E-value = 5e-91 Identities = 181/238(76%) Gaps = 3/238(1%)
It is therefore important: When you report the results of the BLAST search, specify the parameters used for the search allowing the others to reproduce your results.
In contrast to the Nucleotide BLAST, the Protein BLAST calculates the score for matches and mismatches using the pre-defined matrices, PAM and BLOSUM.
PAM (Point Accepted Mutations) matrices (Dayhoff et al., 1978) depict amino acid substitution patterns in 70 groups of closely related (sequence identity > 85%) proteins.
- PAM1 corresponds to 1 aa change per 100 aa (1% divergence).
- PAM100 – 100 changes per 100 aa; made from PAM1.
- PAM250 – 250 changes per 100 aa; made from PAM1.
BLOSUM (BLOcks SUbstitution Matrices) matrices (Henikoff & Henikoff, 1992) depict amino acid substitution in 2,000 blocks (corresponding to structural or functional motifs) in 500 groups of related proteins.
- BLOSUM62 represent the conservation level of sequences used to derive the matrix (seq. identity >62%).
- BLOSUM90 ~ 90%.
- BLOSUM45 ~ 45%.
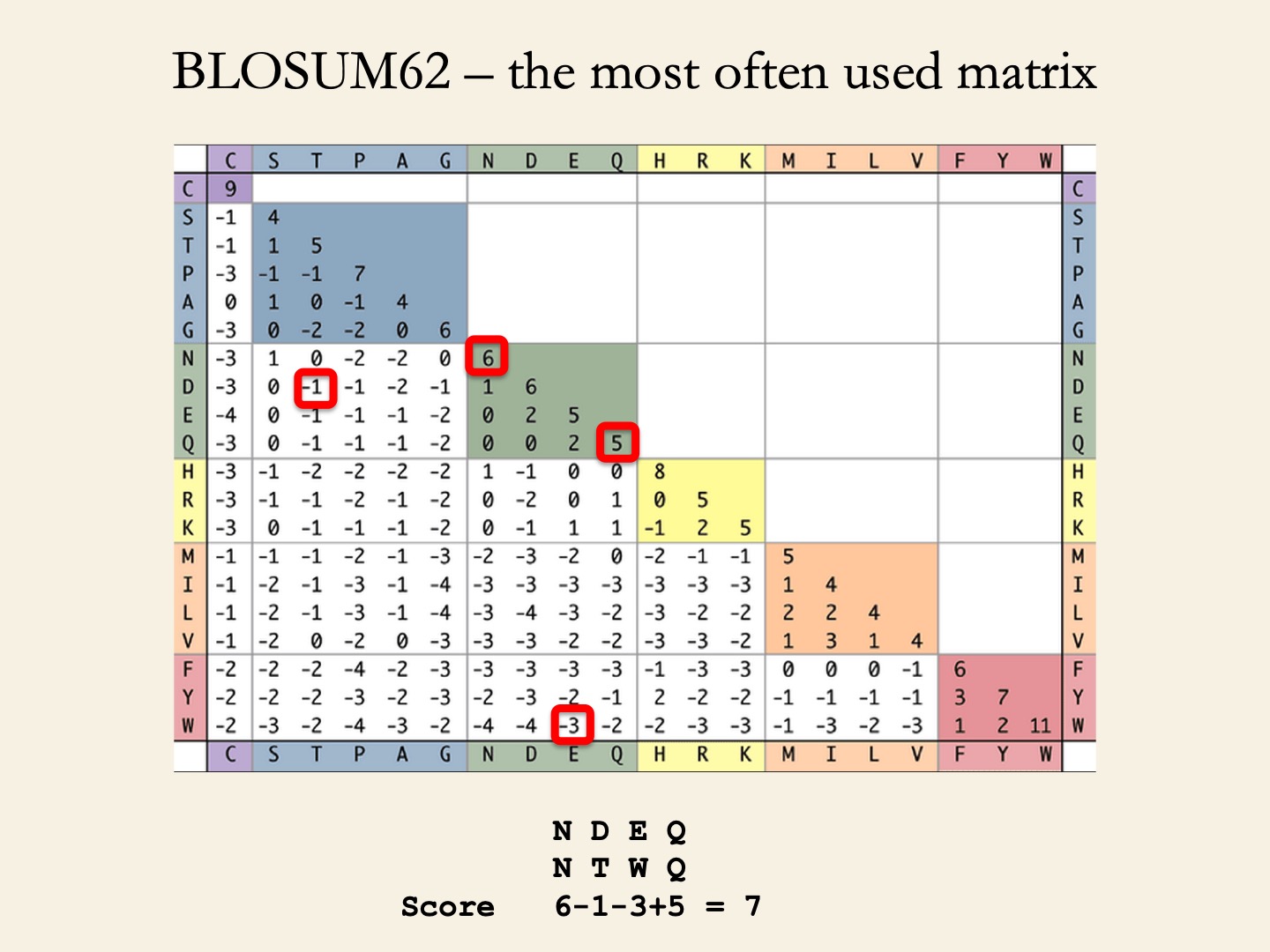
Which matrices to use?
- PAM30 and BLOSUM90 – For short highly significant alignments (above 70-90% sequence identity).
- BLOSUM62 – Detecting members of a protein family (above 50-60% sequence identity).
- PAM250 and BLOSUM45 – For longer alignments of divergent sequences (below 50% sequence identity).
That misterious Expect value, or E-value
- E-value of the alignment is the expected number of sequences that give the same
Z-scoreor better if the database is probed with a random sequence. For example, an E-value of 1 assigned to a hit can be interpreted as meaning that in a database of the current size one might expect to see 1 match with a similar score simply by chance. Z-scoreof thescore Sof the alignment is calculated as(S – mean score over database)/SD.- E-value is not the probability!* If
Pis the probability to obtain by chance an alignment with the sameZ-scoreor better, then P and E (E-value) are related asP = 1- e^(-E). - E-value depends on the size of the database and it ranges from 0 to the number of sequences in the databases.
- The default E-value used by NCBI BLAST is 10.
- The lower the E-value, or the closer it is to zero, the more “significant” the match is.
EXERCISE 3: We saw that the alignment of these two proteins had ~90% similarity (without masking low-complexity regions).
BAG32277
NP_001265185
Q. How will the Blast score and E-value change if the BLOSUM90 matrix is used instead of the default BLOSUM62?
How to infer function of a protein domain
The suggested cut-offs to infer biologically significant similarity using BLAST:
-
For proteins, to predict protein domain 3D structure, use blastp against Protein Data Bank proteins (PDB database): sequence identity > 25% (for protein domains longer 100 aa), E-value < 1e-4.
-
For proteins, to predict protein domain function in absence of structural information, use blastp against annotated proteins (UniProtKB/SwissProt database: sequence identity > 40% (for protein domains longer 100 aa) or higher , depending on function (e.g., 70-80% for enzymes), E-value < 1e-4.
-
For proteins, to identify the protein domain family, use blastp against non-redundant protein sequences (nr): : sequence identity > 25% (for protein domains longer 100 aa), E-value < 1e-4.
-
For nucleotides: sequence identity >= 70% (for the sequence of more than 300 nt), E-value < 1e-6.
Let’s explore domains of the human Homeobox protein ESX1
-
On NCBI website, search for this protein and then on the right panel select “Identify conserved domains”.
-
In UniProt, search for this protein ESX1_HUMAN –> click “Domains and repeats” and Click “View protein in Interpro”.
EXERCISE 4:
Let’s investigate in the protein ESX1_HUMAN the 15 X 9 AA tandem repeats of sequence pattern P-P-x-x-P-x-P-P-x
- Q.1. What are the positions of this region?
- Q.2. Check InterPro on what is known about this region. Was the 3D structure of this region predicted or solved?
Let’s investigate if this 15 X 9 AA tandem repeats of sequence pattern P-P-x-x-P-x-P-P-x is present in other human proteins.
Click BLAST near this region 244 - 378. Following the suggested cut-offs (above), choose the E-value < 0.0001 and human for the Target database, and search the sequence of the 244-378 region against UniProtKB Human database.
- Q.2. How many Reviewed proteins have been found? This is the number of curated human proteins in which this repeat is present.
Now, we can look at a specific alignment. For example, click on View alignment for FMN2_HUMAN - Formin-2 Homo sapiens (Human).
- Q.3. What are positions of this region in the formin-2 protein? Write it down.
- Q.4. Check InterPro if the 3D structure of this region was predicted or solved.
Multiple sequence alignment
“pairwise alignments whispers… multiple alignment shouts out loud” (Hubbard et al., 1996)
Multiple sequence alignment is used to:
- Find structural similarity in proteins and RNA.
- Predict secondary structure and model a protein 3D structure.
- Build phylogenetic trees.
- Annotate a protein sequence to a family or a domain.
- Build a profile for a new family.
- Identify functional motif in DNA or protein.
Popular methods:
- Clustal Omega - implemented on the UniProt website.
- T-Coffee (developed by Cedric Notredame, CRG).
- MUSCLE
- MUFFT
Multiple protein alignment in UniProt
At UniProt, let’s search for Il1a protein.
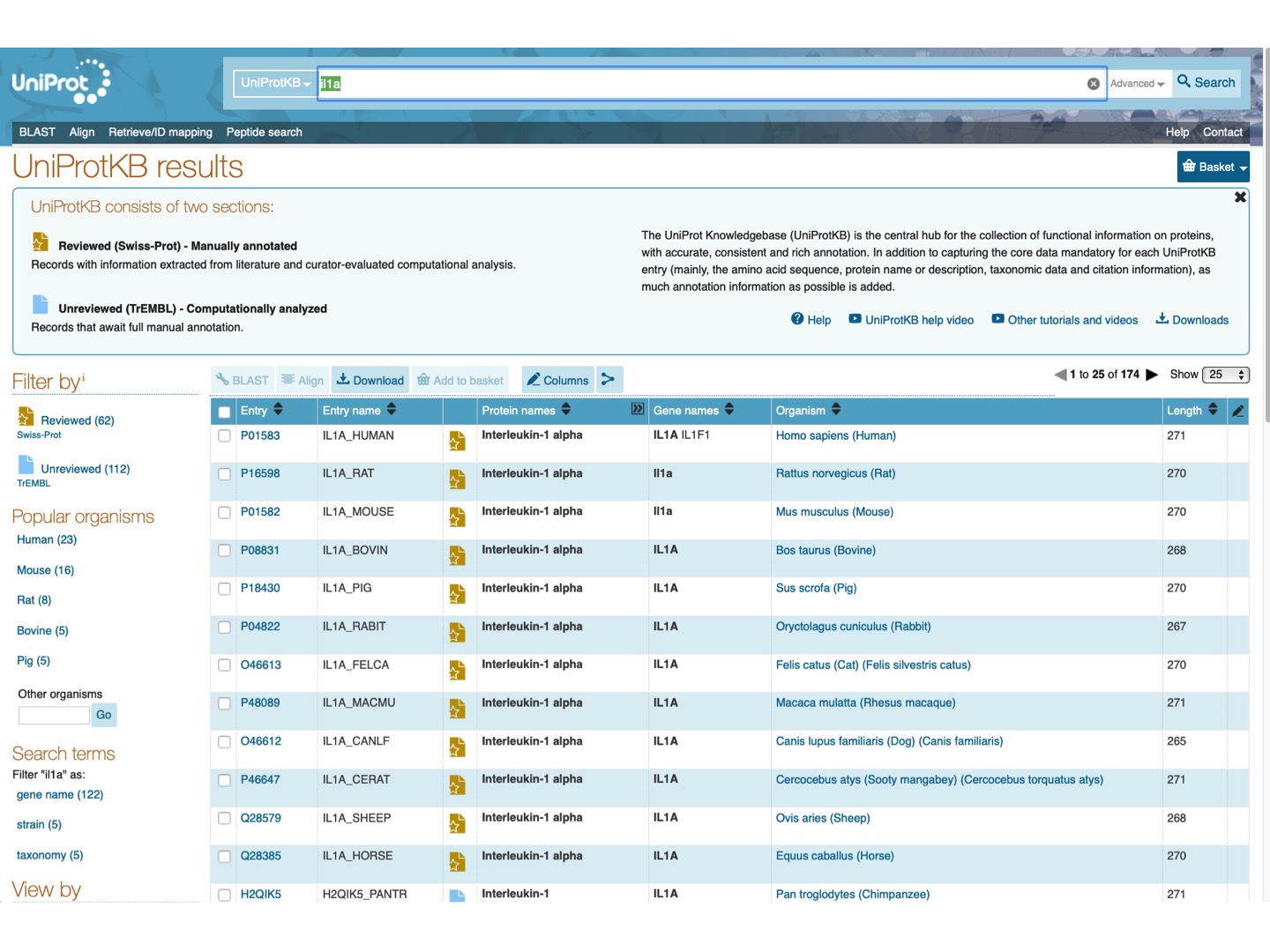
Select Filter by Reviewed and check Il1a protein for a few mammalian species (any 6-15). Click Align on the upper tab.
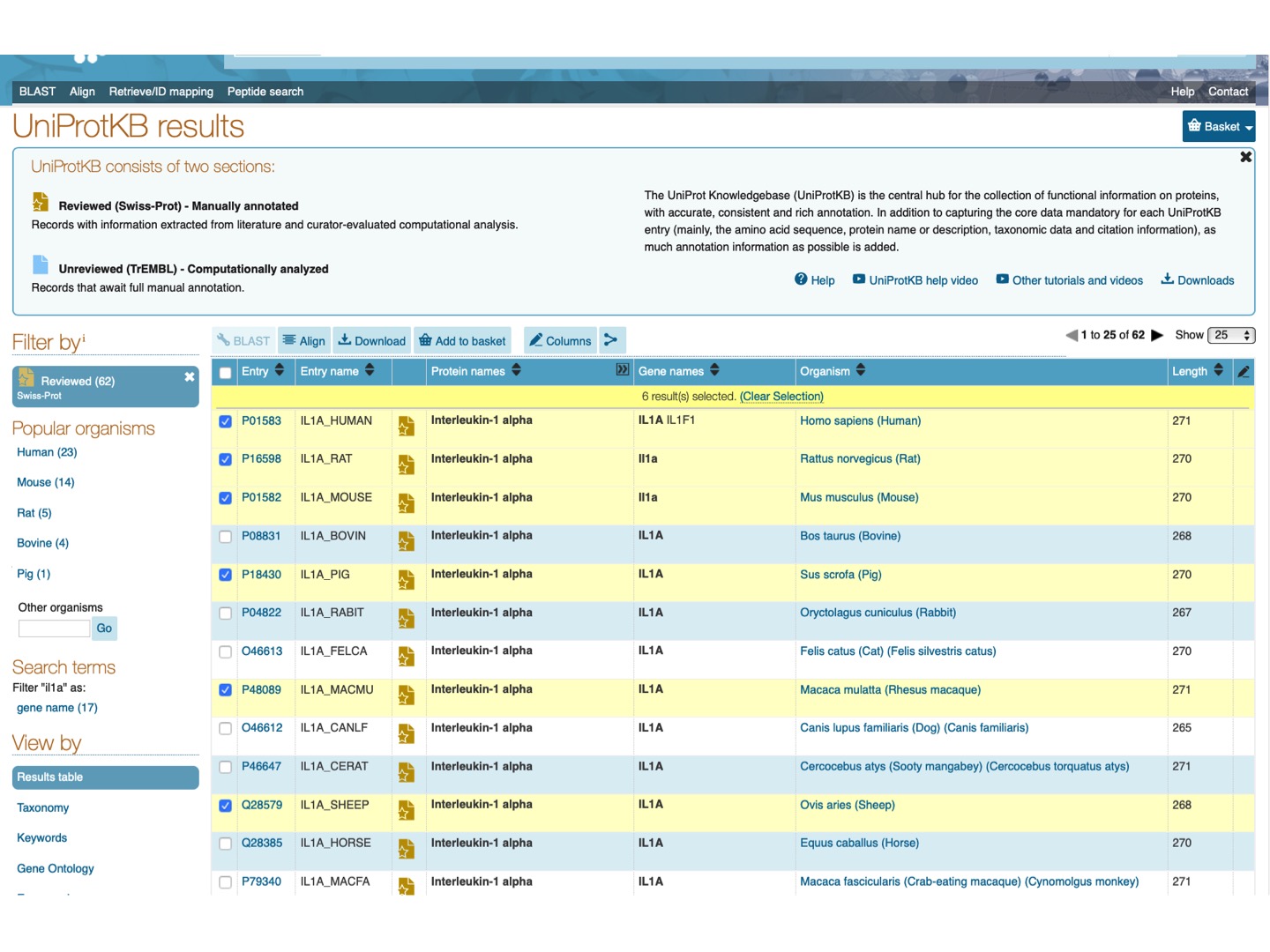
Here is the resulting alignment. You can scroll down or click on the upper left panel to see the phylogenetic tree for the selected species.

Pro-peptide of IL-1α has a functional nucleus localization signal (NLS) with a sequence
LKKRRLS
Can you find this signal and tell if it is conserved in the aligned proteins? (TIP: highlight positive & charged amino acids, as K (Lysine) and R (Arginine) are both positively charged amino acids.)