Hands-on: Linux refresher¶
Linux history¶
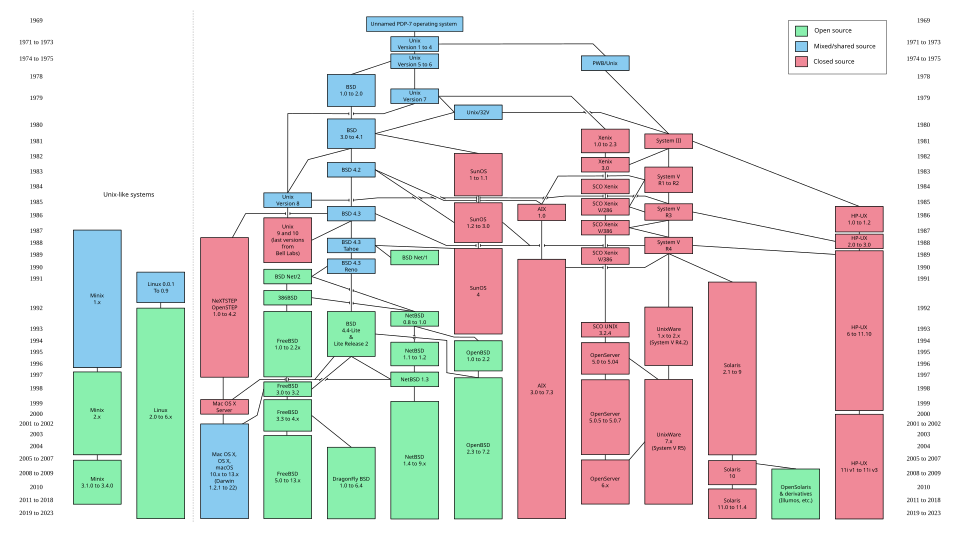
UNIX is a computer operating system originally developed in 1969 by a group of AT&T employees at Bell Labs.
UNIX-like derivatives spread from that moment:
BSD
AIX
HP-UX
Linux
etc.
UNIX Philosophy¶
Portable: Same code should work the same in different machines
Multi-tasking: Different processes can run simultaneously. Every process has a unique identifier (PID)
Multi-user: Many people can use the same machine at the same time. Users can share resources and processes
Use of plain-text for storing data: also for configuration files
Hierarchical file system
Almost everything is a file: That includes devices and some information of processes
Use of small programs all together to retrieve an output instead of an only multifunctional one
Filesystem structure. Linux and Mac¶
/ (Root)
├── bin (Binaries)
├── boot (Boot files)
├── dev (Device files)
├── etc (Configuration files)
├── home (User personal data)
├── lib (Shared libraries)
├── media (Removable media)
├── mnt (Mount directory)
├── opt (Optional software)
├── proc (Process & kernel files)
├── root (Root user home)
├── sbin (System binaries)
├── srv (Service data)
├── tmp (Temporary files)
├── usr (User binaries & data)
│ ├── bin
│ ├── sbin
│ ├── lib
│ └── share
└── var (Variable data)
└── log
You can inspect it with ls /. We will discover more later…
Other UNIX systems (e.g., Mac OS X) are similar, but not exactly the same.
/ (Root)
├── Applications/ (User apps like Chrome, Spotify)
├── Library/ (Shared system resources & settings)
├── System/ (Core macOS files; read-only)
│ └── Applications/ (Built-in apps like Safari, Mail)
├── Users/ (Equivalent to /home in Linux)
│ ├── yourusername/ (Desktop, Documents, etc.)
│ └── Shared/ (Files accessible to all users)
├── Volumes/ (Mounted drives, USBs, and DMGs)
├── bin/ (Essential Unix binaries) -> [Hidden]
├── sbin/ (System administration binaries) -> [Hidden]
├── usr/ (User binaries, libraries, and include files) -> [Hidden]
├── dev/ (Device files) -> [Hidden]
├── private/ (Contains actual etc, var, and tmp) -> [Hidden]
│ ├── etc/ (System configuration files)
│ ├── var/ (Variable data, logs, caches)
│ └── tmp/ (Temporary files)
└── cores/ (Core dumps for debugging) -> [Hidden]
Move around¶
We use what we name the Command Line Interface (CLI) Shell, in our case Bash, as an interface with the Operating System.
Relative paths¶
cd-> go to homecd ../-> go one level backcd Desktop-> go to Desktopcd ../../-> go two levels backcd ../Desktop/..-> don’t movecd .-> go nowhere - ‘.’ means current location
Absolute paths¶
cd /-> go to rootcd /home/training/Desktopcd ~-> go to home
Play with ls and different modifiers: ls -l, ls -lt, ls -la, etc.
Note
Sometimes we get lost and would like to know where we are. We can use the command pwd
Create files and directories¶
To create files and folders in Linux is quite simple. You can use a number of programs for creating an empty file (touch) or an empty directory (mkdir)
touch my_beautiful_file.txt
mkdir my_beautiful_folder
To display the list of files and folder we can use the command ls
ls
my_beautiful_file.txt my_beautiful_folder
To change the name of a file (or a directory) you can use the command mv while for copying the file you can use cp. Adding the option -r (recursive) to cp allows to copy a whole folder and its content.
mv my_beautiful_file.txt my_ugly_file.txt
mv my_beautiful_folder my_ugly_folder
cp my_ugly_file.txt my_beautiful_file.txt
cp -r my_ugly_folder my_beautiful_folder
If you omit the -r option the system will complain
cp my_ugly_folder my_other_folder
cp: omitting directory ‘my_ugly_folder’
You can use mv also for moving a file (or a directory) inside a folder. Also cp will allow you to make a copy inside a folder.
mv my_beautiful_file.txt my_beautiful_folder
cp my_ugly_file.txt my_ugly_folder
ls
my_beautiful_folder my_ugly_file.txt my_ugly_folder
For entering in a folder we can use the tool cd
cd my_ugly_folder
ls
my_ugly_file.txt
For going out we can move one level out
cd ../
ls
my_beautiful_folder my_ugly_file.txt my_ugly_folder
We can write to a file using the character >, that means output redirection.
echo "ATGTACTGACTGCATGCATGCCATGCA" > my_dna.txt
And display the content of the file using the program cat
cat my_dna.txt
ATGTACTGACTGCATGCATGCCATGCA
To convert this sequence to a RNA one we can just replace the T base with U by using the program sed. The syntax of this program is the following s/<TO BE REPLACED>/<TO REPLACE>/.
You can add a g at the end if you want to replace every character found s/<TO BE REPLACED>/<TO REPLACE>/g.
sed 's/T/U/g' my_dna.txt > my_rna.txt
cat my_rna.txt
AUGUACUGACUGCAUGCAUGCCAUGCA
Let’s remove something:
rm my_rna.txt
Let’s try to remove the previous directory:
rm my_beautiful_folder
# We cannot :O
rmdir my_beautiful_folder
# We cannot either
rm my_beautiful_folder/* && rmdir my_beautiful_folder
# Alternative
rm -r my_beautiful_folder # Recursive! Use with care
We can provide access to files and directories installed in other locations to more convenient places (e.g., for performing analyses) and so we save some space. Be careful sometimes if using shared storage.
ln -s my_dna.txt your_dna.txt
Recap¶
touchwrites empty filesmkdircreated empty directoriesmvmove files (or directory) or change their namelslist files and directoriescpcopy files and directoriescdchange the directorypwddisplays where you areechoprint values to standard outputcatprint the content of a file to standard outputsedreplace a string with another
rmremove a filermdirremove an empty directorylnprovides links to files/directories in other locations
Download files¶
Then we can go back to our command line and use the program wget to download that file and using CTRL+C to paste the address:
wget ftp://ftp.ensemblgenomes.org/pub/bacteria/release-42/fasta/bacteria_22_collection/escherichia_coli_bl21_gold_de3_plyss_ag_/dna/README
Define a specific filename.
wget -O my_readme.txt ftp://ftp.ensemblgenomes.org/pub/bacteria/release-42/fasta/bacteria_22_collection/escherichia_coli_bl21_gold_de3_plyss_ag_/dna/README
Capture log (stdout and stderr)
wget -O my_readme.txt ftp://ftp.ensemblgenomes.org/pub/bacteria/release-42/fasta/bacteria_22_collection/escherichia_coli_bl21_gold_de3_plyss_ag_/dna/README > wget.log 2>&1
A similar existing tool is curl:
curl -o my_readme.txt ftp://ftp.ensemblgenomes.org/pub/bacteria/release-42/fasta/bacteria_22_collection/escherichia_coli_bl21_gold_de3_plyss_ag_/dna/README > curl.log 2>&1
curl is more powerful and you can do more stuff than simply download files (e.g., test API services)
Piping¶
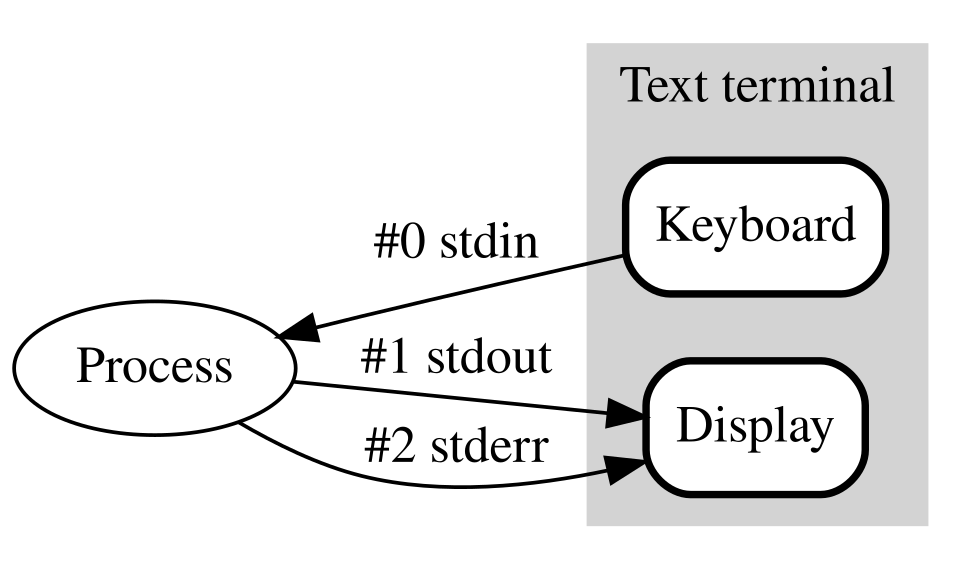
Pipe stdout to another command:
ls | grep ".txt"
Lists files, then filters for .txt files.
Redirect stdout to a file (overwrite):
echo "Hello" > output.txt
Append stdout to a file:
echo "World" >> output.txt
Redirect stderr to a file:
ls non_existing_file 2> error.log
Redirect both stdout and stderr to a file:
command > all_output.log 2>&1
# Use the wget or curl example above
Basic commands for manipulating text files¶
We will use some of them during the course in practical examples.
more/lesspaginate contents of a file if it is large andcatis not convenienttarcan be used for pack and unpack several files in a single archivegzip/gunzipare used for compressing and uncompressing files - normally resulting in files with.gzextensionzcatis likecatbut allowing to view gzipped filesgrepfinds patternscutsplits the contents, normally in a line-by-line basisheadprints the starting contentstailprints the ending contentssortsorts the content of a file or stdinwccounts words, characters, or lines
Below a couple of examples:
# Let's download a gzipped FASTQ file
wget https://biocorecrg.github.io/RNAseq_coursesCRG_2026/latest/data/reads/SRR3091420_1_chr6.fastq.gz
# We see its contents without uncompressing it
zcat SRR3091420_1_chr6.fastq.gz
# We pipe it to the paginator
zcat SRR3091420_1_chr6.fastq.gz | less
# We look for 55 within the file
zcat SRR3091420_1_chr6.fastq.gz | grep 55
# We look for 55 within the file and we read interactively
zcat SRR3091420_1_chr6.fastq.gz | grep 55 | less
# We get the first 10 lines of the file
zcat SRR3091420_1_chr6.fastq.gz | head -n 10
# We get the last 10 lines of the file
zcat SRR3091420_1_chr6.fastq.gz | tail -n 10
# We uncompress the file
gunzip SRR3091420_1_chr6.fastq.gz
# We paginate the uncompressed file
less SRR3091420_1_chr6.fastq
# We compress it back
gzip SRR3091420_1_chr6.fastq
# Let's download a tar archive
wget https://biocorecrg.github.io/RNAseq_coursesCRG_2026/latest/data/annotation/reference_chr6_Hsapiens.tar.gz
# Let's inspect its contents
tar tf reference_chr6_Hsapiens.tar.gz
# reference_chr6/
# reference_chr6/Homo_sapiens.GRCh38.dna.chrom6.fa.gz
# reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf.gz
# reference_chr6/gencode.v49.transcripts.chr6.fa.gz
# Let's extract a specific file
tar xf reference_chr6_Hsapiens.tar.gz reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf.gz
# Let's inspect its last 10 lines
zcat reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf.gz | tail -n 10
# Let's extract the fourth column of the 100 last lines and we place the contents into a file
zcat reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf.gz | tail -n 100 | cut -f 4 > start-points.txt
# Let's inspect that file and we sort is contents
less start-points.txt
sort start-points.txt | less
sort start-points.txt | wc -l
sort -u start-points.txt | less
sort -u start-points.txt | wc -l
Running programs¶
Making and Running a Bash Script¶
Create a file called
myscript.shwith this content:echo 'echo "Hello, world!"' > myscript.sh
Make it executable:
chmod +x myscript.sh
Run it:
./myscript.sh
Finding running programs¶
When you run a program in Linux, the shell searches for its executable in directories listed in the PATH environment variable. You can check your current PATH with:
echo $PATH
$PATH is one of the many available environment variables. You can check all them with env.
If you want to add a directory to your PATH permanently, for example bin subdirectory in your $HOME directory, edit your .bashrc file (in your home directory) and add:
export PATH="$PATH:~/bin"
After saving .bashrc, reload it with:
source ~/.bashrc
Environment variables like PATH control how programs are found and executed. Modifying .bashrc lets you customize your shell environment.
We can test it placing myscript.sh in the new PATH folder we defined:
mv myscript.sh ~/bin
Running containers¶
We will use already pre-made containers, so we will not need installing so many programs (and struggle with their dependencies).
For executing the tools from the container images, we will use Apptainer/Singularity.
Why Apptainer/Singularity?
Not as popular as Docker, but very convenient for Bioinformatics and HPC environments
You don’t need to worry so much about different kind of UNIX permissions
For the command-line part of the course not happening in RStudio, we will use this container image we generated with Seqera Containers.
singularity pull RNAseq_course.sif docker://community.wave.seqera.io/library/fastq-screen_fastqc_kraken2_multiqc_pruned:3161b0b514b51263
singularity exec -e RNAseq_course.sif fastqc --version
Along the course we will use this shortcut for no needing to type so much:
# Important: RNASeq_course.sif must be in home ~
export RUN="singularity exec -e $HOME/RNAseq_course.sif"
$RUN fastqc --version
Further recommendations¶
You can browse the contents of a directory in a Graphical User Interface (GUI) calling the file explorer program and the target directory.
# This opens Nautilus - GUI file explorer in the current directory
# Used in Ubuntu GNOME environment
nautilus .
There are also many CLI text editors available (e.g., vim, nano, etc.), but we can also choose to use GUI editors.
By default, by using open <file> command, we will open the actual file with the default application associated with that file type.
open myfile.txt
We expect that text files in the Ubuntu GNOME environment will be opened with: gnome-text-editor.
Tip
If you execute open <directory>, it will actually open that directory with the default file explorer.
# Open current directory in a file explorer
open .
See also
In Ubuntu, default applications can also be changed for specific file types following these instructions: https://help.ubuntu.com/stable/ubuntu-help/files-open.html.en