Hands-on: Read mapping¶
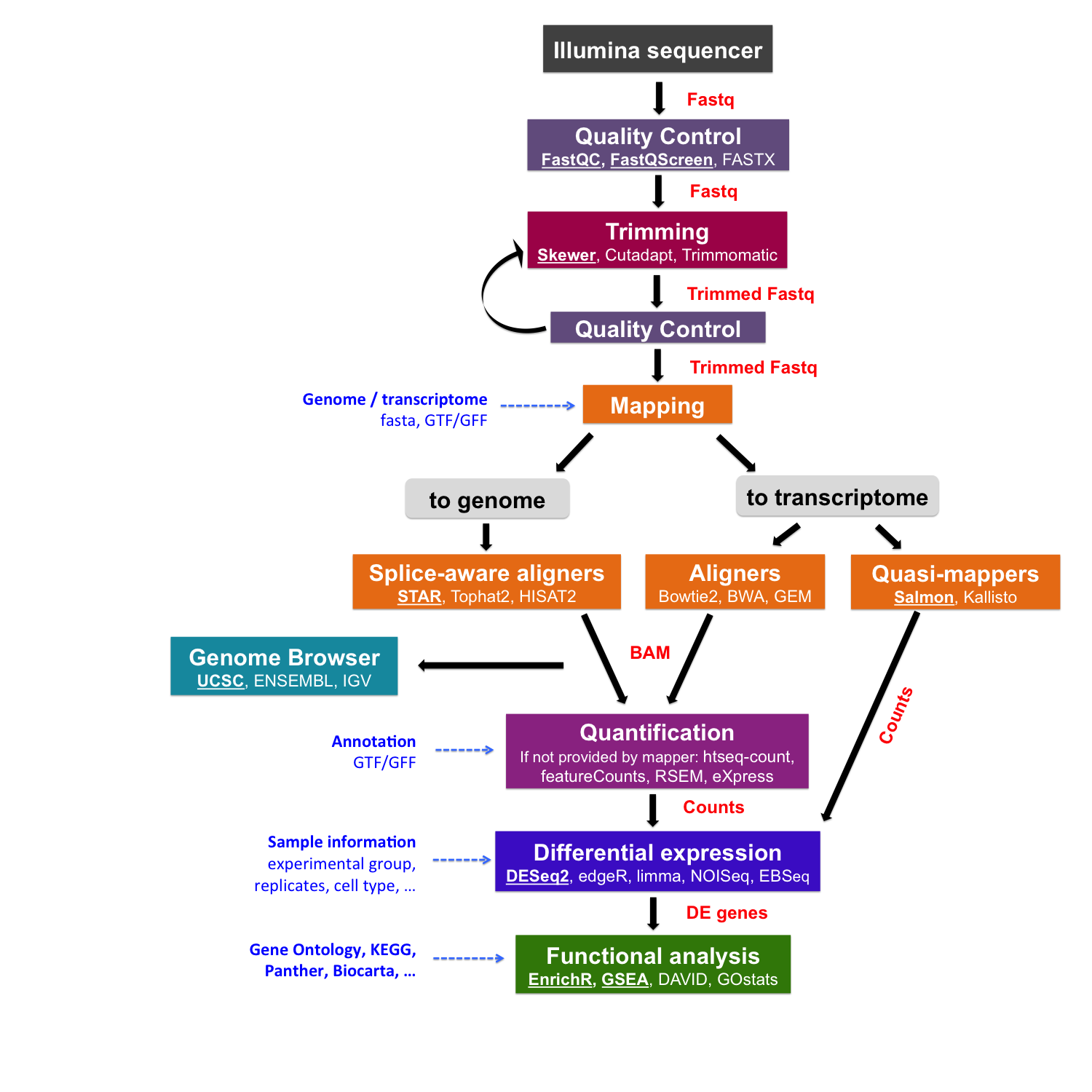
What does it mean to map reads to a transcriptome? During sequencing, we read both ends of each RNA fragment—these are called “paired-end reads.” Mapping to the transcriptome means finding where these paired reads match in our database of known transcript sequences. Since transcripts already have introns removed and exons joined together, the reads align directly without needing to “jump” across gaps as they would when mapping to the genome.
|
From wikimedia commons |
Tools for read mapping¶
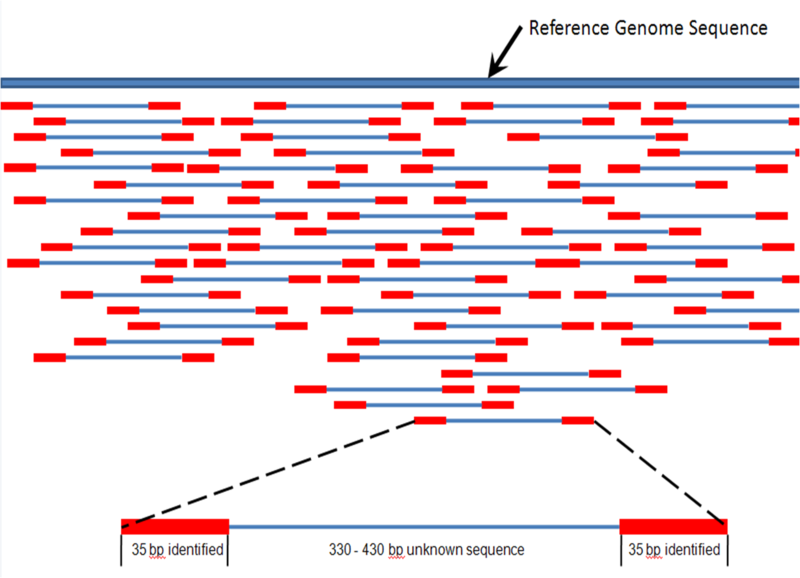
Multiples aligners were developed over the last decades, using different algorithms:
Read mappers timeline |
|---|
|
Nuno A. Fonseca, Johan Rung, Alvis Brazma, John C. Marioni, Tools for mapping high-throughput sequencing data, Bioinformatics, Volume 28, Issue 24, December 2012, Pages 3169–3177, https://doi.org/10.1093/bioinformatics/bts605 |
Index¶
Before doing the mapping, we have to prepare an index from the reference DNA sequence that a chosen algorithm will use.
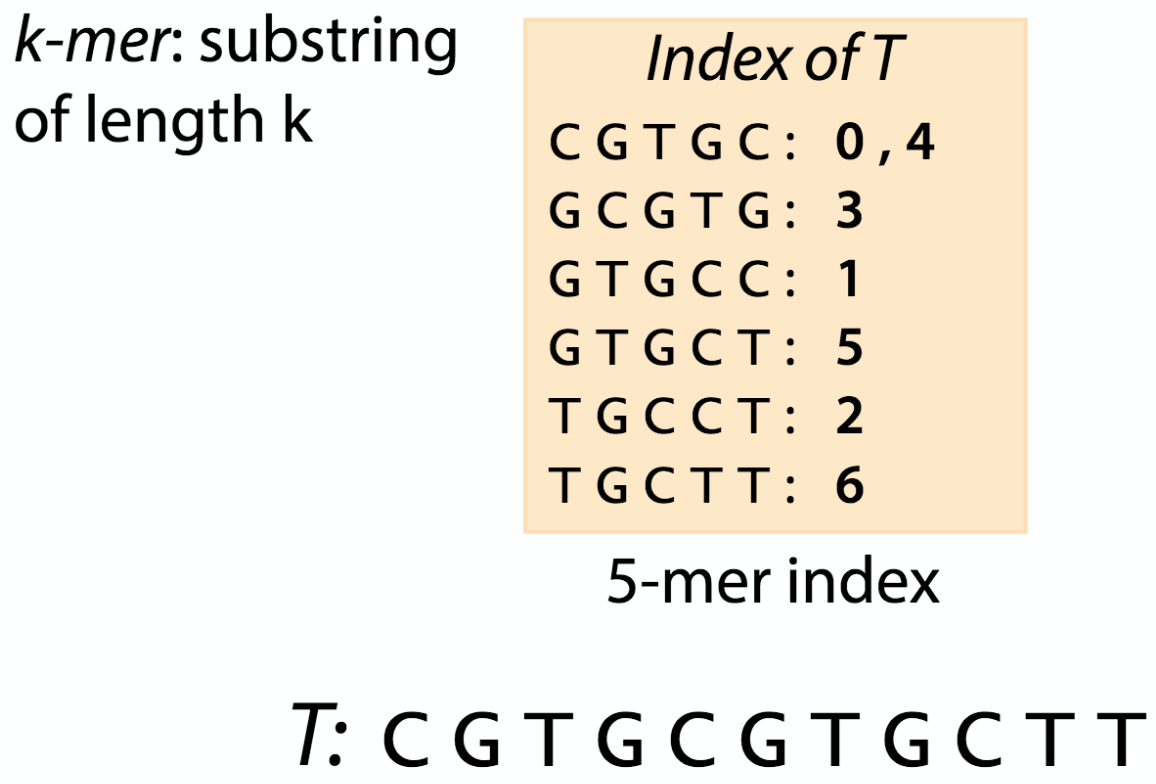
Like the index at the end of a book, an index of a large DNA sequence allows one to rapidly find shorter sequences embedded in it. Different tools use different approaches at genome/transcriptome indexing.
k-mer index |
|---|
|
from coursera.org |
Fast (splice-unaware) aligners to a reference transcriptome¶
These tools can be used for aligning short reads to a transcriptome reference.
If a genome were used as a reference, these tools would not map reads to splicing junctions.
They can be much faster than traditional aligners like Blast but less sensitive and may have limitations about the read size.
Bowtie is an ultrafast, memory-efficient short read aligner geared toward quickly aligning large sets of short DNA sequences (reads) to large genomes/transcriptomes. Bowtie uses a Burrows-Wheeler index.
Bowtie2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. It is particularly good at aligning reads of length 50 up to 100s or 1,000s to relatively long (e.g. mammalian) genomes. Bowtie 2 indexes the transcriptome with an FM Index.
BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. BWA indexes the genome with an FM Index.
Splice-aware aligners to a reference genome¶
These aligners are able to map to the splicing junctions described in the annotation and even to detect novel ones.
Some of them can detect gene fusions and SNPs and also RNA editing. For some of these tools, the downstream analysis requires the assignation of the aligned reads to a given gene/transcript.
HISAT2 is the next generation of spliced aligner from the same group that have developed TopHat. It is a fast and sensitive alignment program for mapping next-generation sequencing reads (both DNA and RNA) to a population of human genomes (as well as to a single reference genome). The indexing scheme is called a Hierarchical Graph FM index (HGFM).
STAR is an ultrafast universal RNA-seq aligner. It uses sequential maximum mappable seed search in uncompressed suffix arrays followed by seed clustering and stitching procedure. It is also able to search for gene fusions.
Quasi-mappers (alignment-free mappers) to a reference transcriptome¶
These tools are way faster than the previous ones because they don’t need to report the resulting alignments (BAM/SAM files) but only associate a read to a given transcript for quantification. They don’t discover novel transcript variants (or splicing events) or detect variations, etc.
Salmon is an advanced version of Sailfish, by the same authors, tool for wicked-fast transcript quantification from RNA-seq data. It requires a set of target transcripts to quantify and a K-mer parameter to make the index (i.e. minimum acceptable alignment).
Kallisto is a program for quantifying abundances of transcripts from bulk and single-cell RNA-Seq data. It is based on the novel idea of pseudoalignment for rapidly determining the compatibility of reads with targets, without the need for alignment.
Before proceeding, we need to retrieve a reference genome or transcriptome from a public database, along with its annotation:
A FASTA file contains the actual genome/transcriptome sequence.
A GTF/GFF file contains the corresponding annotation.
We already have them here:
# go to the appropriate folder
cd ~/rnaseq_course/reference_genome
# download reference files for chromosome 6
wget https://biocorecrg.github.io/RNAseq_coursesCRG_2026/latest/data/annotation/reference_chr6_Hsapiens.tar.gz
# extract archive
tar -xvzf reference_chr6_Hsapiens.tar.gz
# remove remaining .tar.gz archive
rm reference_chr6_Hsapiens.tar.gz
Mapping using STAR¶
For the STAR running options, see STAR Manual.
Building the STAR index¶
To make an index for STAR, we need both the genome sequence in FASTA format and the annotation in GTF format.
As STAR is very resource-consuming, we will create an index for chromosome 6 only, and let’s hope it will work! :).
However, STAR requires unzipped .fa and .gtf files. We need to unzip them.
# go to reference_genome folder
cd ~/rnaseq_course/reference_genome/reference_chr6
# unzip files (keep original zipped file)
zcat Homo_sapiens.GRCh38.115.chr6.gtf.gz > Homo_sapiens.GRCh38.115.chr6.gtf
zcat Homo_sapiens.GRCh38.dna.chrom6.fa.gz > Homo_sapiens.GRCh38.dna.chrom6.fa
Q. How much (in percentage) disk space is saved when those two files are kept zipped vs unzipped?
Once the index is built, do not forget to remove those unzipped files!
To index the genome with STAR for RNA-seq analysis, the sjdbOverhang option needs to be specified for detecting possible splicing sites:
It usually equals the minimum read size minus 1; it tells STAR what is the maximum possible stretch of sequence that can be found on one side of a splicing site.
In our case, since the read size is 49 bases, we can accept a maximum of 48 bases on one side and one base on the other of a splicing site; that is, to set up this parameter to 48.
This also means that for every different read-length to be aligned, a new STAR index needs to be generated. Otherwise, a drop in aligned reads can be experienced.
--runThreadNallows you to parallelize the job.
NOTE that for small genomes, parameter --genomeSAindexNbases (default 14) should be scaled down as: min(14, log2(GenomeLength)/2 - 1). Here: min(14, log2(170805979/2)-1) =~ 12.6
Building the STAR index (option --runMode genomeGenerate):
# make a mapping folder
mkdir cd ~/rnaseq_course/mapping
# go to mapping folder
cd ~/rnaseq_course/mapping
# create a sub-folder where the index will be generated
mkdir index_star_chr6
# remember to have defined the environmental variable RUN to access your singularity image
# We use the RNAseq_course.sif image we downloaded in previous sections
export RUN="singularity exec -e $HOME/RNAseq_course.sif"
# create the index and store it in ~/rnaseq_course/mapping/index_star_chr6
$RUN STAR --runMode genomeGenerate --genomeDir index_star_chr6 \
--genomeFastaFiles ~/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.dna.chrom6.fa \
--sjdbGTFfile ~/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf \
--sjdbOverhang 48 \
--genomeSAindexNbases 12.6 \
--outFileNamePrefix Hsapiens_chr6 \
--runThreadN 1
--genomeSAindexNbases: default 14. If the genome is small, it should be scaled down as: min(14, log2(GenomeLength)/2 - 1). Here: min(14, log2(170805979/2)-1) =~ 12.6
This should take around 3 to 4 minutes to complete.
Aligning reads to the genome¶
To use STAR for the read alignment (default --runMode option), we have to specify the following options:
the index directory (
--genomeDir)the read files (
--readFilesIn)if reads are compressed or not (
--readFilesCommand)
The following parameters are optional but very convenient:
--outSAMtype: type of output. Default is BAM Unsorted; STAR outputs unsorted Aligned.out.bam file(s). “The paired ends of an alignment are always adjacent, and multiple alignments of a read are adjacent as well. This ”unsorted” file cannot be indexed or directly used with downstream software such as HTseq, without the need for sorting.” We therefore prefer the option BAM SortedByCoordinate--outFileNamePrefix: the path for the output directory and prefix of all output files. By default, this parameter is ./, i.e. all output files are written in the current directory.--quantMode. With the--quantMode GeneCountsoption set, STAR will count the number of reads per gene while mapping. A read is counted if it overlaps (1nt or more) one and only one gene. In the case of mapping paired-end data, both ends are checked for overlaps. The counts coincide with those produced by htseq-count with default parameters. This option requires annotations (in GTF format or GFF with–-sjdbGTFfileoption) used at the genome generation step, or at the mapping step. (from STAR Manual)
We can try to launch the mapping for one file:
# go to mapping folder
cd ~/rnaseq_course/mapping
# create sub-folder where we will store the alignments
mkdir alignments_STAR
# we place trimmed reads in a location that is convenient
# as written below, index_star_chr6 must be in the same location as you are running the program
$RUN STAR --genomeDir index_star_chr6 \
--readFilesIn ~/rnaseq_course/trimmed_reads/SRR3091420_1_chr6_trimmed.fq.gz \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--quantMode GeneCounts \
--outFileNamePrefix alignments_STAR/SRR3091420_1_chr6 \
--runThreadN 1
If this was successful and not too slow and resource-consuming, you can do it for all samples, in a loop:
for fastq in ~/rnaseq_course/trimmed_reads/*_trimmed.fq.gz
do echo $fastq
$RUN STAR --genomeDir index_star_chr6 \
--readFilesIn $fastq \
--readFilesCommand zcat \
--outSAMtype BAM SortedByCoordinate \
--quantMode GeneCounts \
--outFileNamePrefix alignments_STAR/$(basename $fastq _trimmed.fq.gz)
done
BACKUP !!
If it was indeed too resource-consuming, you can download the aligned files in BAM format from:
cd ~/rnaseq_course/mapping
# get alignments and indexes ending with bam or bai
wget -r -np -nH --cut-dirs=5 -A "*.bam*" https://biocorecrg.github.io/RNAseq_coursesCRG_2026/latest/data/aln
# extract the counts and the logs
tar -xvzf all.logs.tar.gz
tar -xvzf all.tabs.tar.gz
Let’s explore the output directory “alignments” or the backup folder.
ls -lh alignments
Inspecting the log file can give you a hint about the quality of the mapping:
cat SRR3091420_1_chr6Log.final.out
Started job on | Mar 09 17:57:58
Started mapping on | Mar 09 17:57:59
Finished on | Mar 09 17:58:07
Mapping speed, Million of reads per hour | 375.83
Number of input reads | 835168
Average input read length | 48
UNIQUE READS:
Uniquely mapped reads number | 786944
Uniquely mapped reads % | 94.23%
Average mapped length | 48.38
Number of splices: Total | 91355
Number of splices: Annotated (sjdb) | 90653
Number of splices: GT/AG | 90810
Number of splices: GC/AG | 431
Number of splices: AT/AC | 15
Number of splices: Non-canonical | 99
Mismatch rate per base, % | 0.20%
Deletion rate per base | 0.00%
Deletion average length | 1.59
Insertion rate per base | 0.00%
Insertion average length | 1.17
MULTI-MAPPING READS:
Number of reads mapped to multiple loci | 46554
% of reads mapped to multiple loci | 5.57%
Number of reads mapped to too many loci | 98
% of reads mapped to too many loci | 0.01%
UNMAPPED READS:
Number of reads unmapped: too many mismatches | 0
% of reads unmapped: too many mismatches | 0.00%
Number of reads unmapped: too short | 1570
% of reads unmapped: too short | 0.19%
Number of reads unmapped: other | 2
% of reads unmapped: other | 0.00%
CHIMERIC READS:
Number of chimeric reads | 0
% of chimeric reads | 0.00%
Read counts¶
STAR outputs read counts per gene into PREFIXReadsPerGene.out.tab file with 4 columns which correspond to different strandedness options:
column 1 |
gene ID |
|---|---|
column 2 |
counts for unstranded RNA-seq |
column 3 |
counts for the 1st read strand aligned with RNA (htseq-count option -s yes) |
column 4 |
counts for the 2nd read strand aligned with RNA (htseq-count option -s reverse) |
Let’s see what the ReadsPerGene.out.tab file looks like for sample SRR3091420_1_chr6:
head SRR3091420_1_chr6ReadsPerGene.out.tab
gene id |
read counts per gene (unstranded) |
read counts per gene (read 1) |
read counts per gene (read 2) |
|---|---|---|---|
N_unmapped |
1670 |
1670 |
1670 |
N_multimapping |
46554 |
46554 |
46554 |
N_noFeature |
24060 |
376484 |
397091 |
N_ambiguous |
74169 |
21118 |
17326 |
ENSG00000219375 |
1 |
1 |
0 |
ENSG00000270174 |
0 |
0 |
0 |
ENSG00000261730 |
0 |
0 |
0 |
ENSG00000176515 |
0 |
0 |
0 |
ENSG00000286368 |
0 |
0 |
0 |
ENSG00000217239 |
31 |
22 |
9 |
Select the output according to the strandedness of your data.
Note, if you have stranded data and choose one of the columns 3 or 4, the other column (4 or 3) will give you the count of antisense reads.
For example, in the stranded protocol shown in “Library preparation”, Read 1 is mapped to the antisense strand (this is also true for single-end reads), while Read 2, to the sense strand.
Which protocol, stranded or unstranded, was used for this RNA-seq data?
We can count the number of reads mapped to each strand by using a simple awk script:
grep -v "N_" SRR3091420_1_chr6ReadsPerGene.out.tab | awk '{unst+=$2;forw+=$3;rev+=$4}END{print unst,forw,rev}'
688715 389342 372527
# 688715 389342 372527
We see that 389,342 Reads 1 (forward) were mapped to known genes and 372,527 Reads 2 (reverse) were mapped to known genes.
These numbers are very similar, which indicates that the protocol used for this mRNA-Seq experiment is unstranded.
If the protocol used was stranded, there would be a strong imbalance between the number of reads mapped to known genes in forward versus reverse strands.
BAM/SAM/CRAM format¶
The BAM format is a compressed version of the SAM format (which is a plain text) and cannot thus being seen as a text. To explore the BAM file, we have to convert it to the SAM format by using samtools. Note that we use the parameter -h to show also the header that is hidden by default.
$RUN samtools view -h SRR3091420_1_chr6Aligned.sortedByCoord.out.bam | head -n 10
@HD VN:1.4 SO:coordinate
@SQ SN:6 LN:170805979
@PG ID:STAR PN:STAR VN:2.7.11b CL:/opt/conda/bin/STAR-avx2 --genomeDir index_star_chr6 --readFilesIn /users/bi/lcozzuto/rnaseq_course/trimmed_reads/SRR3091420_1_chr6_trimmed.fq.gz --readFilesCommand zcat --outFileNamePrefix alignments_STAR/SRR3091420_1_chr6 --outSAMtype BAM SortedByCoordinate --quantMode GeneCounts
@PG ID:samtools PN:samtools PP:STAR VN:1.17 CL:/usr/local/bin/samtools view -h SRR3091420_1_chr6Aligned.sortedByCoord.out.bam
@CO user command line: /opt/conda/bin/STAR-avx2 --genomeDir index_star_chr6 --readFilesIn /users/bi/lcozzuto/rnaseq_course/trimmed_reads/SRR3091420_1_chr6_trimmed.fq.gz --readFilesCommand zcat --outSAMtype BAM SortedByCoordinate --quantMode GeneCounts --outFileNamePrefix alignments_STAR/SRR3091420_1_chr6
10416098 0 6 113167 255 48M * 0 0 GGGAAAAGTACAAATTCAACATGTAATTGTATAGTAATCCATATAAAA bbbeeeeecggggiiiiiiiiiihhhiiighhiihhhhigiiiiiiii NH:i:1 HI:i:1 AS:i:47 nM:i:0
8553177 272 6 119288 3 48M * 0 0 GAAATCCAGTGGGACAGTCAAATCTTAAAGCTCCAAAATGATCTCCTT iiiiiiiiiiiiigiiiiiiiihiihiiiiihhiigggggeeeeebbb NH:i:2 HI:i:2 AS:i:47 nM:i:0
4630026 272 6 128432 3 49M * 0 0 AGCACTAACCATTGTAGCATGCCAATATACTCAAAATTCAATGAAATTC hfgehhggiihhhiiihhiihhhhffffghdihhiifggggeeeeebbb NH:i:2 HI:i:2 AS:i:48 nM:i:0
10689795 0 6 135934 255 49M * 0 0 AAGGCTGCAATGAGCTGTGATCGCACCACCGCACCCAAGCCTGGGTGGT bbbeeeeeggggfiiiighiiiiiiiiiiiiiihiihfhhiiiii_ega NH:i:1 HI:i:1 AS:i:44 nM:i:2
10416101 0 6 136561 255 48M * 0 0 CCCAACGTTTAGACTACACAATGAGTTAAGAACGACAAAAATAAGCTC ___ecccceeeeghhhhhhhhgfgiihfhhhhfhffffgghhhidfff NH:i:1 HI:i:1 AS:i:47 nM:i:0
The first part indicated by the first character @ in each row is the header:
Tag |
Field |
Value |
|---|---|---|
@HD |
VN |
1.4 |
@HD |
SO |
coordinate |
@SQ |
SN |
6 |
@SQ |
LN |
170805979 |
@PG |
ID |
STAR |
@PG |
PN |
STAR |
@PG |
VN |
2.7.11b |
@PG |
CL |
|
@PG |
ID |
samtools |
@PG |
PN |
samtools |
@PG |
PP |
STAR |
@PG |
VN |
1.17 |
@PG |
CL |
|
@CO |
- |
user command line: |
The rest is a read alignment.
Field |
Value |
|---|---|
Read ID |
10416098 |
FLAG |
0 |
Chromosome |
6 |
Position |
113167 |
Mapping Quality |
255 (uniquely mapped) |
CIGAR |
48M (48 bp match) |
Sequence |
|
Quality |
|
* FLAG 0 means that the read is mapped on the forward strand.
CIGAR string 48M means that 48 bases were mapped to the reference (M).
You can use this website for the translation of SAM FLAG values and this one for interpreting CIGAR strings.
Extra fields are often present and differ between aligners [https://samtools.github.io/hts-specs/SAMtags.pdf](https://samtools.github.io/hts-specs/SAMtags.pdf). In our case we have:
Field |
Meaning |
|---|---|
NH:i:1 |
number of mappings to the reference |
HI:i:1 |
which alignment is the reported one (in this case is the second one) |
AS:i:47 |
Alignment score calculated by the aligner |
nM:i:0 |
number of difference with the reference* |
Note
Careful that sometimes tools can disagree on some definition, from time to time, the standard is “changed” depending on the community, new data to show
Let’s convert BAM to SAM:
$RUN samtools view -h SRR3091420_1_chr6Aligned.sortedByCoord.out.bam > SRR3091420_1_chr6Aligned.sortedByCoord.out.sam
You can see that the SAM file is 5 times larger than the BAM file.
Yet, the more efficient way to store the alignment is to use the CRAM format. CRAM is fully compatible with BAM, and main repositories, such as GEO and SRA, accept alignments in the CRAM format. UCSC Genome Browser can visualize both BAM and CRAM files. It is now a widely recommended format for storing alignments.
To convert BAM to CRAM, we have to provide an unzipped and indexed version of the genome.
$RUN samtools faidx ~/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.dna.chrom6.fa
$RUN samtools view -C SRR3091420_1_chr6Aligned.sortedByCoord.out.bam -T ~/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.dna.chrom6.fa -o SRR3091420_1_chr6Aligned.sortedByCoord.out.cram
You can see that a .cram file is twice as small as a .bam file.
ls SRR3091420_1_chr6Aligned.sortedByCoord.out.* -alht
.rw-r----- lcozzuto Bioinformatics_Unit 15 MB Mon Mar 9 19:27:00 2026 SRR3091420_1_chr6Aligned.sortedByCoord.out.cram
.rw-r----- lcozzuto Bioinformatics_Unit 139 MB Mon Mar 9 19:26:05 2026 SRR3091420_1_chr6Aligned.sortedByCoord.out.sam
.rw-r----- lcozzuto Bioinformatics_Unit 27 MB Mon Mar 9 18:58:07 2026 SRR3091420_1_chr6Aligned.sortedByCoord.out.bam
Alignment QC¶
The quality of the resulting alignment can be checked using the tool QualiMap. To run QualiMap, we specify the kind of analysis (rnaseq), the gtf file, and the strandness of the library (-p unstranded).
Note that if the library was paired-end, you would add the -pe option*.
IMPORTANT: before running QualiMap ensure enough disk space for a temporary directory ./tmp that the program is required, running the following command:
export _JAVA_OPTIONS="-Djava.io.tmpdir=./tmp -Xmx6G"
cd ~/rnaseq_course/mapping
# create folder
mkdir qc_qualimap
# run qualimap
$RUN qualimap rnaseq -bam SRR3091420_1_chr6Aligned.sortedByCoord.out.bam \
-gtf ~/rnaseq_course/reference_genome/reference_chr6/Homo_sapiens.GRCh38.115.chr6.gtf \
-outdir qc_qualimap \
-p non-strand-specific
We can check the final report in a browser:
firefox qc_qualimap/qualimapReport.html
If you cannot, you can reach it here
The report gives a lot of useful information, such as the total number of mapped reads, the number of reads mapped to exons, introns, or intergenic regions, and the bias towards one of the ends of mRNA (which can give information about RNA integrity or the protocol used).
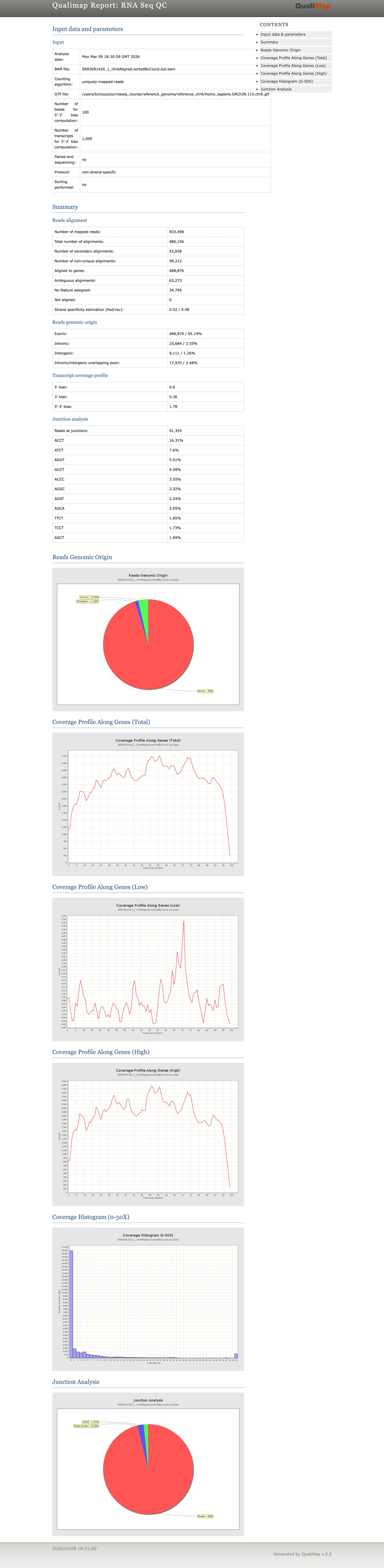
Finally, we can see that the majority of reads map to the exons.
IMPORTANT for running QualiMap on many samples (for detail, see QualiMap documentation
Make sure to give to the output folder the name corresponding to a running sample; e.g., ./QC/SRR3091420_1_chr6; otherwise, output files will be overwritten.
If you run QualiMap in parallel for many samples, make sure to create a different tmp-folder for each sample; e.g., ./tmp/SRR3091420_1_chr6Aligned.
QualiMap sorts BAM files by read names. To speed up this part of the program execution, you can use samtools to sort the BAM files in parallel and using multiple CPUs, and then give QualiMap a BAM file sorted by read names and provide an option –sorted.
Genome Browser¶
To explore BAM and CRAM files produced by the STAR mapper, we first need to sort and index the files. In our case, sorting has already been done by STAR’s BAM SortedByCoordinate option.
The indexing can be done with samtools:
cd ~/rnaseq_course/mapping
$RUN samtools index SRR3091420_1_chr6Aligned.sortedByCoord.out.bam
IMPORTANT!
Be careful with the chromosome name conventions!
Different genome browsers name chromosomes differently. UCSC names chromosomes as chr1, chr2,…chrM; while Ensembl, 1, 2, … MT.
When you map reads to a genome with a given convention, you cannot directly display BAM/CRAM files in a genome browser that uses a different convention.
GENCODE uses the UCSC convention, while ENSEMBL doesn’t: we need to change the chromosome names before being able to load them in the UCSC Genome Browser.
cd ~/rnaseq_course/mapping
# create new sub-directory
mkdir bam_ucsc
# convert chromosome naming (produce a SAM file)
$RUN samtools view -h SRR3091420_1_chr6Aligned.sortedByCoord.out.bam | awk -F "\t" 'BEGIN{OFS="\t"}{if($1 ~ /^@/){print $0} else {print $1,$2,"chr"$3,$4,$5,$6,$7,$8,$9,$10,$11,$12}}' | sed 's/chrMT/chrM/g' | sed 's/SN:/SN:chr/g' > SRR3091420_1_chr6Aligned.sam
# convert SAM to BAM
$RUN samtools view -b -o SRR3091420_1_chr6Aligned.bam SRR3091420_1_chr6Aligned.sam
# create index for BAM file
$RUN samtools index SRR3091420_1_chr6Aligned.bam
# remove SAM files
rm *.sam
Mapping using Salmon¶
Salmon is a tool for quantifying the expression of transcripts using RNA-seq data.
It is a quasi-mapper: it doesn’t produce the read alignments (and doesn’t output BAM/SAM files). Salmon “quasi-maps” read to the transcriptome rather than to the genome.
Salmon can also make use of pre-computed alignments (in the form of a SAM/BAM file) instead of the FASTQ files.
Building the Salmon index¶
To make an index for Salmon, we need transcript sequences in the FASTA format.
This can be found easily in GENCODE.
The transcript sequences corresponding to chromosome 6 were prepared and already downloaded in ~/rnaseq_course/reference_genome/
Salmon does not need any decompression of the input, so we can index by using this command:
cd ~/rnaseq_course/mapping
# index and store the index files in the index_salmon folder
$RUN salmon index -t ~/rnaseq_course/reference_genome/reference_chr6/gencode.v49.transcripts.chr6.fa.gz \
-i index_salmon \
--gencode
We add the parameter --gencode as our data come from Gencode and their header contains several identifiers separated by the character |. This parameter allows the program to parse the header and keep only the transcript identifier.
But as we commented in the previous presentation, Salmon works better by avoiding the misplacement of DNA to actual transcripts. So we create a “joint” reference by concatenating transcripts, and the genome and a decoy.txt file with the list of sequences to e considered as decoy.
cd ~/rnaseq_course/reference_genome/reference_chr6/
cp gencode.v49.transcripts.fa.gz gencode.v49.transcripts.genome.fa.gz
cat Homo_sapiens.GRCh38.dna.chrom6.fa.gz >> gencode.v49.transcripts.genome.fa.gz
echo "6" > decoy.txt
Then, we index again
cd ~/rnaseq_course/mapping
# index and store the index files in the index_salmon folder. More threads are needed; it is quite slow!
$RUN salmon index -t ~/rnaseq_course/reference_genome/reference_chr6/gencode.v49.transcripts.genome.fa.gz \
-i index_salmon \
--gencode -p 4 -d decoy.txt
This step takes around 10 minutes with 4 cpus.
Quantifying transcript expression¶
To quantify reads with Salmon, we need to specify the type of sequencing library, aka Fragment Library Types in Salmon, using three letters:
The first:
Symbol |
Meaning |
Reads |
|---|---|---|
I |
inward |
-> … <- |
O |
outward |
<- … -> |
M |
matching |
-> … -> |
The second:
Symbol |
Meaning |
|---|---|
S |
stranded |
U |
unstranded |
The third:
Symbol |
Meaning |
|---|---|
F |
read 1 (or single-end read) comes from the forward strand |
R |
read 1 (or single-end read) comes from the reverse strand |
From the STAR output for read counts we already know that for the analyzed experiment, the U (Unstranded) library was used.
If the library were paired-end and sequenced with a stranded reverse library, we would set the parameter to ISR.
If we want to assign the reads to the genes (option -g) in addition to transcripts, we have to provide a GTF file corresponding to the transcript version that was used to build the Salmon index.
We have it already for chromosome 6, in ~/rnaseq_course/reference_genome/
We can proceed with the mapping.
cd ~/rnaseq_course/mapping
# create folder for salmon's output files
mkdir alignments_salmon
# again here using more cpus is better
$RUN salmon quant -i index_salmon -l U \
-r ~/rnaseq_course/raw_data/fastq_chr6/SRR3091420_1_chr6.fastq.gz \
-o alignments_salmon/SRR3091420_1_chr6_salmon \
--seqBias \
--validateMappings -p 4
We can check the results inside the folder “alignments”.
ls alignments_salmon/SRR3091420_1_chr6_salmon/
For an explanation of all output files, see the Salmon documentation.
The most interesting to us in this course is the file quant.sf, which is a tab-separated file containing the read counts for transcripts:
awk '{if ($4>0) print}' quant.sf| head
Name Length EffectiveLength TPM NumReads
ENST00000466430.5 2748 2815.573 0.384361 1.091
ENST00000424587.7 5603 6195.161 0.635870 3.972
ENST00000743878.1 737 509.108 11.858146 6.087
ENST00000416931.1 372 226.773 5.674886 1.298
ENST00000457540.1 1044 1378.147 1.891150 2.628
ENST00000427426.1 682 558.366 1.979782 1.115
ENST00000514057.1 681 678.963 4.016001 2.749
ENST00000487214.1 865 562.716 1.762567 1.000
ENST00000959612.1 3051 4260.652 0.232787 1.000
We will use information on read counts for transcripts from quant.sf files for the differential expression (DE) analysis.
Now, if time and resources allow, proceed with the mapping of the 9 remaining samples:
for fastq in ~/rnaseq_course/ ~/rnaseq_course/trimmed_reads/SRR309142*.gz;
do echo $fastq
$RUN salmon quant -i index_salmon -l U \
-r ${fastq} \
-o alignments_salmon/$(basename $fastq _trimmed.fq.gz)_salmon \
--seqBias \
--validateMappings;
done
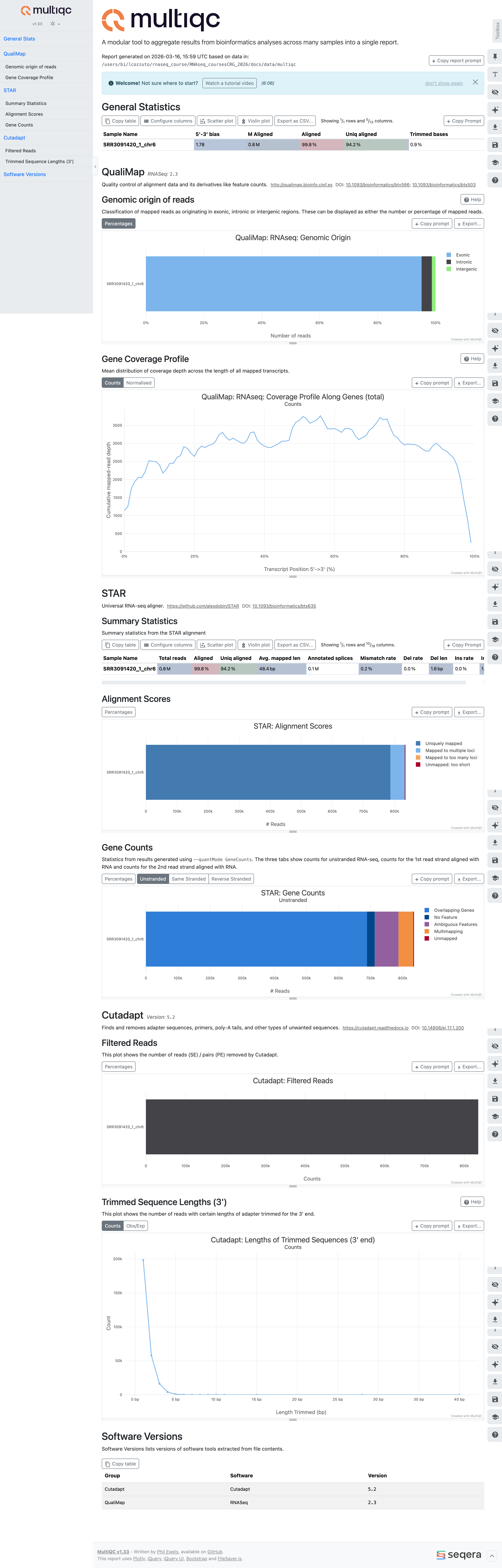
MultiQC report¶
At this point, we can summarize all the work done with the tool MultiQC. MultiQC aggregates outputs from many bioinformatics tools across many samples into a single report by searching a given directory for analysis logs and compiling an HTML report.
MultiQC supports many tools: see the different modules
Let’s create a multiqc_report folder and link all the analyses done so far.
cd ~/rnaseq_course/
# create a folder for the multiqc result
mkdir multiqc_report
cd ~/rnaseq_course/multiqc_report
# link QC, trimmed_reads and mapping data
ln -s ~/rnaseq_course/quality_control* .
ln -s ~/rnaseq_course/mapping* .
ln -s ~/rnaseq_course/trimmed_reads
Then run multiqc on the directory multiqc_report to combine all reports:
cd ~/rnaseq_course/multiqc_report
$RUN multiqc .
/// MultiQC 🔍 v1.33
file_search | Search path: /users/bi/lcozzuto/rnaseq_course/RNAseq_coursesCRG_2026/docs/data/multiqc
searching | ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 40/40
qualimap | Found 1 RNASeq reports
star | Found 1 reports and 1 gene count files
cutadapt | Found 1 reports
write_results | Data : multiqc_data
write_results | Report : multiqc_report.html
multiqc | MultiQC complete
We can visualize the final report in the internet browser:
firefox multiqc_report.html
Here is an example done on just one sample