Hands-on: Running the full analysis using the nf-core pipelines¶
All the previous steps can be chained together in what bioinformaticians call a pipeline. Historically, each lab developed its own pipelines using custom code tailored to its specific IT infrastructure. In many cases, this code was so ad hoc that adapting it to other environments was nearly impossible. This was a major barrier to reproducibility and collaboration. This challenge sparked some open-source projects, including one invented here at CRG: Nextflow.
Nextflow is a domain-specific language built on Groovy that enables scalable, reproducible, and portable scientific workflows. It abstracts away infrastructure complexity, allowing the same pipeline to run on a laptop, HPC cluster, or cloud environment. From the very beginning, a community of scientists adopted the tool and began building a curated collection of high-quality, peer-reviewed bioinformatics pipelines. This community, known as nf-core, now maintains over 100 pipelines covering diverse applications—from RNA-seq and variant calling to metagenomics and spatial transcriptomics. All nf-core pipelines follow strict guidelines for structure, documentation, and testing, ensuring consistency and reliability across the entire collection.
These pipelines leverage containerization to ensure reproducibility and portability. Each bioinformatics tool runs inside a Linux container (such as Singularity/Apptainer or Docker), eliminating dependency conflicts and the “works (only :) ) on my machine” problem. Most nf-core pipelines rely on container images from Biocontainers, a community-driven project that automatically builds and hosts containers for thousands of bioinformatics tools. These pre-built images are publicly available at quay.io, allowing pipelines to pull the exact software versions they need on demand.
The nf-core community has a great documentation, which is being updated continuously.
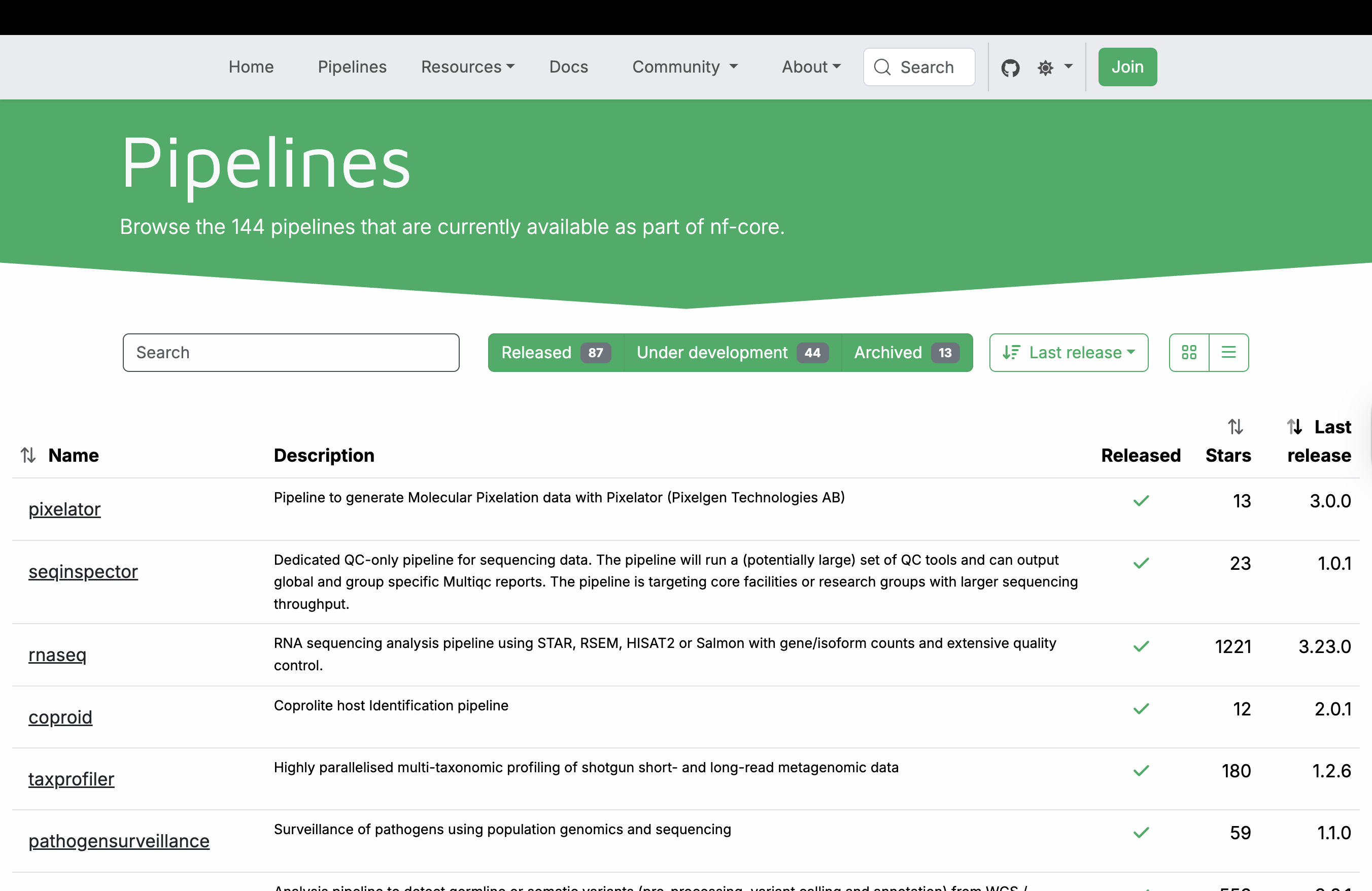
RNA-seq nf-core pipeline¶
For our data analysis, we will use the nf-core RNA-seq pipeline. The whole pipeline is described in this fancy diagram:

As you can see, we have almost 40 tools that are chained and available. This level of complexity was nearly impossible in the past without the use of an orchestrator.
We can identify 5 macro areas:
Preprocessing¶
Merge different sequencing runs, infer strandness, QC, UMI extraction, adapter trimming and contaminants, and rRNA removal.
Genome alignment¶
Genome alignment using either Star, Hisat2 or bowtie2. Quantification with Rsem or Salmon. (Hisat2 has no quantification method)
Transcriptome pseudo-alignment¶
Transcriptome pseudo-alignment with either Salmon or Kallisto.
Post-processing after genome alignment¶
Sorting, umi and read deduplication, transcriptome assembly, and quantification. Generation of read coverage files (bigWig) for displaying in genome browsers.
Quality control and reporting after post-processing¶
QC on alignment, such as RSeQC, qualimap, etc. Detection of contamination and final reporting with MultiQC
We can use the nf-core tools by installing them using pip.
Note
You might also think of getting nf-core tools from Biocontainers, but this won’t work when executing the pipeline!
# -r indicates the exact version to fetch
nf-core pipelines download nf-core/rnaseq -r dev
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 3.5.2 - https://nf-co.re
WARNING Could not find GitHub authentication token. Some API requests may fail.
In addition to the pipeline code, this tool can download software containers.
? Download software container images: (Use arrow keys)
» none
singularity
docker
In this way, you can also pre-download the containers. This can be useful if your HPC has no internet connection and needs to run the pipeline offline.
In addition to the pipeline code, this tool can download software containers.
? Download software container images: none
If transferring the downloaded files to another system, it can be convenient to have everything compressed in a single file.
? Choose compression type: (Use arrow keys)
» none
tar.gz
tar.bz2
zip
After that, a new folder named nf-core-rnaseq_dev appears with the whole pipeline. You can navigate inside and see all the files:
ls nf-core-rnaseq_dev/dev/
tests
subworkflows
ro-crate-metadata.json
nf-test.config
nextflow_schema.json
nextflow.config
modules
modules.json
main.nf
docs
conf
bin
assets
README.md
LICENSE
CODE_OF_CONDUCT.md
CITATIONS.md
CHANGELOG.md
You don’t need to change anything except the file base.config inside conf folder. Inside are described the resources needed for the different processes, and some of them can be really too generous.
/*
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
nf-core/rnaseq Nextflow base config file
~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
A 'blank slate' config file, appropriate for general use on most high performance
compute environments. Assumes that all software is installed and available on
the PATH. Runs in `local` mode - all jobs will be run on the logged in environment.
----------------------------------------------------------------------------------------
*/
process {
// TODO nf-core: Check the defaults for all processes
cpus = { 1 * task.attempt }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
errorStrategy = { task.exitStatus in ((130..145) + 104 + 175) ? 'retry' : 'finish' }
maxRetries = 1
maxErrors = '-1'
// Process-specific resource requirements
// NOTE - Please try and reuse the labels below as much as possible.
// These labels are used and recognised by default in DSL2 files hosted on nf-core/modules.
// If possible, it would be nice to keep the same label naming convention when
// adding in your local modules too.
// See https://www.nextflow.io/docs/latest/config.html#config-process-selectors
withLabel:process_single {
cpus = { 1 }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_low {
cpus = { 2 * task.attempt }
memory = { 12.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_medium {
cpus = { 6 * task.attempt }
memory = { 36.GB * task.attempt }
time = { 8.h * task.attempt }
}
withLabel:process_high {
cpus = { 12 * task.attempt }
memory = { 72.GB * task.attempt }
time = { 16.h * task.attempt }
}
withLabel:process_long {
time = { 20.h * task.attempt }
}
withLabel:process_high_memory {
memory = { 200.GB * task.attempt }
}
withLabel:error_ignore {
errorStrategy = 'ignore'
}
withLabel:error_retry {
errorStrategy = 'retry'
maxRetries = 2
}
withLabel: process_gpu {
accelerator = 1
containerOptions = { params.gpu_container_options ?: (workflow.containerEngine in ['singularity', 'apptainer'] ? '--nv' : '--gpus all') }
}
}
You can either manually change them or make a new profile and override some of them. Let’s modify the processes with too much RAM or cpus.
process {
// TODO nf-core: Check the defaults for all processes
cpus = { 1 * task.attempt }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
errorStrategy = { task.exitStatus in ((130..145) + 104 + 175) ? 'retry' : 'finish' }
maxRetries = 1
maxErrors = '-1'
// Process-specific resource requirements
// NOTE - Please try and reuse the labels below as much as possible.
// These labels are used and recognised by default in DSL2 files hosted on nf-core/modules.
// If possible, it would be nice to keep the same label naming convention when
// adding in your local modules too.
// See https://www.nextflow.io/docs/latest/config.html#config-process-selectors
withLabel:process_single {
cpus = { 1 }
memory = { 6.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_low {
cpus = { 2 * task.attempt }
memory = { 12.GB * task.attempt }
time = { 4.h * task.attempt }
}
withLabel:process_medium {
cpus = { 6 * task.attempt }
memory = { 24.GB * task.attempt }
time = { 8.h * task.attempt }
}
withLabel:process_high {
cpus = { 8 * task.attempt }
memory = { 48.GB * task.attempt }
time = { 16.h * task.attempt }
}
withLabel:process_long {
time = { 20.h * task.attempt }
}
withLabel:process_high_memory {
memory = { 96.GB * task.attempt }
}
withLabel:error_ignore {
errorStrategy = 'ignore'
}
withLabel:error_retry {
errorStrategy = 'retry'
maxRetries = 2
}
withLabel: process_gpu {
accelerator = 1
containerOptions = { params.gpu_container_options ?: (workflow.containerEngine in ['singularity', 'apptainer'] ? '--nv' : '--gpus all') }
}
}
We need to define the input files inside a sample sheet.
vi sample_sheet.csv
sample,fastq_1,fastq_2,strandedness
SRR3091423,./reads/SRR3091423_1_chr6.fastq.gz,,reverse
SRR3091427,./reads/SRR3091427_1_chr6.fastq.gz,,reverse
SRR3091420,./reads/SRR3091420_1_chr6.fastq.gz,,reverse
SRR3091424,./reads/SRR3091424_1_chr6.fastq.gz,,reverse
SRR3091428,./reads/SRR3091428_1_chr6.fastq.gz,,reverse
SRR3091421,./reads/SRR3091421_1_chr6.fastq.gz,,reverse
SRR3091425,./reads/SRR3091425_1_chr6.fastq.gz,,reverse
SRR3091429,./reads/SRR3091429_1_chr6.fastq.gz,,reverse
SRR3091422,./reads/SRR3091422_1_chr6.fastq.gz,,reverse
SRR3091426,./reads/SRR3091426_1_chr6.fastq.gz,,reverse
Now we can run the wizard:
nf-core pipelines launch nf-core-rnaseq_dev/dev/
,--./,-.
___ __ __ __ ___ /,-._.--~\
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/tools version 3.5.2 - https://nf-co.re
INFO NOTE: This tool ignores any pipeline parameter defaults overwritten by Nextflow config files or profiles
INFO [✓] Default parameters match schema validation
INFO [✓] Pipeline schema looks valid (found 129 params)
INFO Would you like to enter pipeline parameters using a web-based interface or a command-line wizard?
? Choose launch method (Use arrow keys)
Web based
» Command line
We choose the command line, we choose -profile [singularity], as input sample_sheet.csv, outfolder as output. We provide the fasta file as –fasta and the gtf as –gtf.
Note
Hyphens matter! Core Nextflow command-line options use one (-), whereas pipeline-specific parameters use two (–).
Continue >>
INFO [✓] Input parameters look valid
fatal: not a git repository (or any parent up to mount point /)
Stopping at filesystem boundary (GIT_DISCOVERY_ACROSS_FILESYSTEM not set).
INFO Nextflow command:
nextflow run /users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-core-rnaseq_dev/dev -profile "singularity" -params-file
"/users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-params.json"
Do you want to run this command now? [y/n] (y): y
INFO Launching workflow! 🚀
Nextflow 26.02.0-edge is available - Please consider updating your version to it
N E X T F L O W ~ version 25.11.0-edge
Launching `/users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-core-rnaseq_dev/dev/main.nf` [special_lamarck] DSL2 - revision: ff377cb1d2
While the pipeline is running, we can inspect where the pipeline is writing the temporary files: the work folder.
This folder will contain a number of subfolders, that are connected to each process execution. For instance the process indicated with:
[a8/9852eb] Submitted process > NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_FASTQC_UMITOOLS_TRIMGALORE:FASTQC (SRR3091428)
is indicating the existence of a subfolder structure work/a8/9852eb73bcf5c0cebe39fead0be5b5/
Let’s go inside and check what’s there:
ls work/a8/9852eb73bcf5c0cebe39fead0be5b5/
.command.begin
.command.env
.command.err
.command.log
.command.out
.command.run
.command.sh
.command.trace
.exitcode
SRR3091428_raw_fastqc.html
SRR3091428_raw_fastqc.zip
SRR3091428_raw.gz ⇒ SRR3091428_1_chr6.fastq.gz
SRR3091428_1_chr6.fastq.gz ⇒ /users/bi/lcozzuto/rnaseq_course/RNAseq_coursesCRG_2026/docs/data/reads/SRR3091428_1_chr6.fastq.gz
You can see that the input file is a soft link to the original fastq file to avoid unnecessary duplication of data. Moreover, everything that is needed for the computation is “isolated” in this folder without the possibility of file collision with other executions. There is a further link to “standardize” the input name, this is quite useful considering the final reporting with MultiQC.
Inspecting the .command.sh reveal:
#!/usr/bin/env bash -C -e -u -o pipefail
printf "%s %s\n" SRR3091428_1_chr6.fastq.gz SRR3091428_raw.gz | while read old_name new_name; do
[ -f "${new_name}" ] || ln -s $old_name $new_name
done
fastqc \
--quiet \
--threads 2 \
--memory 6144 \
SRR3091428_raw.gz
# capture process environment
...
In brief, the command creates a soft link for standardizing the name and contains the fastqc command line with the definition of memory and the number of threads. This definition is not fixed; it is generated at run time, depending on your configuration, and is linked to the resources requested to your system when submitting the jobs.
Nextflow takes care of all these aspects, so that an increase of resources is automatically translated into changing the command line.
You can also run nextflow without using the nf-core tools. Just using the command line that was written before in the logs or present in the first row of the file .nextflow.log
head -n 1 .nextflow.log
Mar-09 17:07:35.926 [main] DEBUG nextflow.cli.Launcher - $> nextflow run /users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-core-rnaseq_dev/dev -profile singularity -params-file /users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-params.json
In case of failure, you can resume by just adding the -resume parameter to the nextflow command line. You can also change the maximum resources available by using a local config file like
vi my_local.config
process {
resourceLimits = [
cpus: 4,
memory: 6.GB,
time: 24.h
]
}
And then feeding the custom config file using the -c parameter
nextflow run /users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-core-rnaseq_dev/dev -profile singularity -params-file /users/bi/lcozzuto/rnaseq_course/test_nf-core/nf-params.json -c my_local.config -resume
Nextflow versions¶
Nextflow has both stable and edge releases. Some pipelines may require the use of Nextflow edge releases to exploit particular features.
Nextflow installs the latest stable version by default. You can get an edge release either by defining the exact version with NXF_VER or by using the NXF_EDGE environment variable:
NXF_VER=25.11.0-edge nextflow run ...
After some minutes, we got:
[fd/068789] NFCORE_RNASEQ:RNASEQ:BAM_RSEQC:RSEQC_INFEREXPERIMENT (SRR3091426) [100%] 10 of 10 ✔
[d3/c4cf70] NFCORE_RNASEQ:RNASEQ:BAM_RSEQC:RSEQC_JUNCTIONANNOTATION (SRR3091426) [100%] 10 of 10 ✔
[90/34d2d5] NFCORE_RNASEQ:RNASEQ:BAM_RSEQC:RSEQC_JUNCTIONSATURATION (SRR3091426) [100%] 10 of 10 ✔
[b1/4ebfc3] NFCORE_RNASEQ:RNASEQ:BAM_RSEQC:RSEQC_READDISTRIBUTION (SRR3091426) [100%] 10 of 10 ✔
[0d/599b93] NFCORE_RNASEQ:RNASEQ:BAM_RSEQC:RSEQC_READDUPLICATION (SRR3091426) [100%] 10 of 10 ✔
[f1/c62ae3] NFCORE_RNASEQ:RNASEQ:MULTIQC (1) [100%] 1 of 1 ✔
-[nf-core/rnaseq] Pipeline completed successfully -
-[nf-core/rnaseq] Please check MultiQC report: 10/10 samples failed strandedness check.-
Completed at: 09-Mar-2026 17:24:00
Duration : 16m 21s
CPU hours : 1.7
Succeeded : 363
Let’s inspect the output.
fastqc (QC)
raw
trim
fq_lint (validator)
multiqc (reporting)
pipeline_info
star_salmon: output of several tools:
bam files
salmon quantification on star aligned results
qualimap QC
…
trimgalore (report of trimgalore execution)
The final report can be seen here
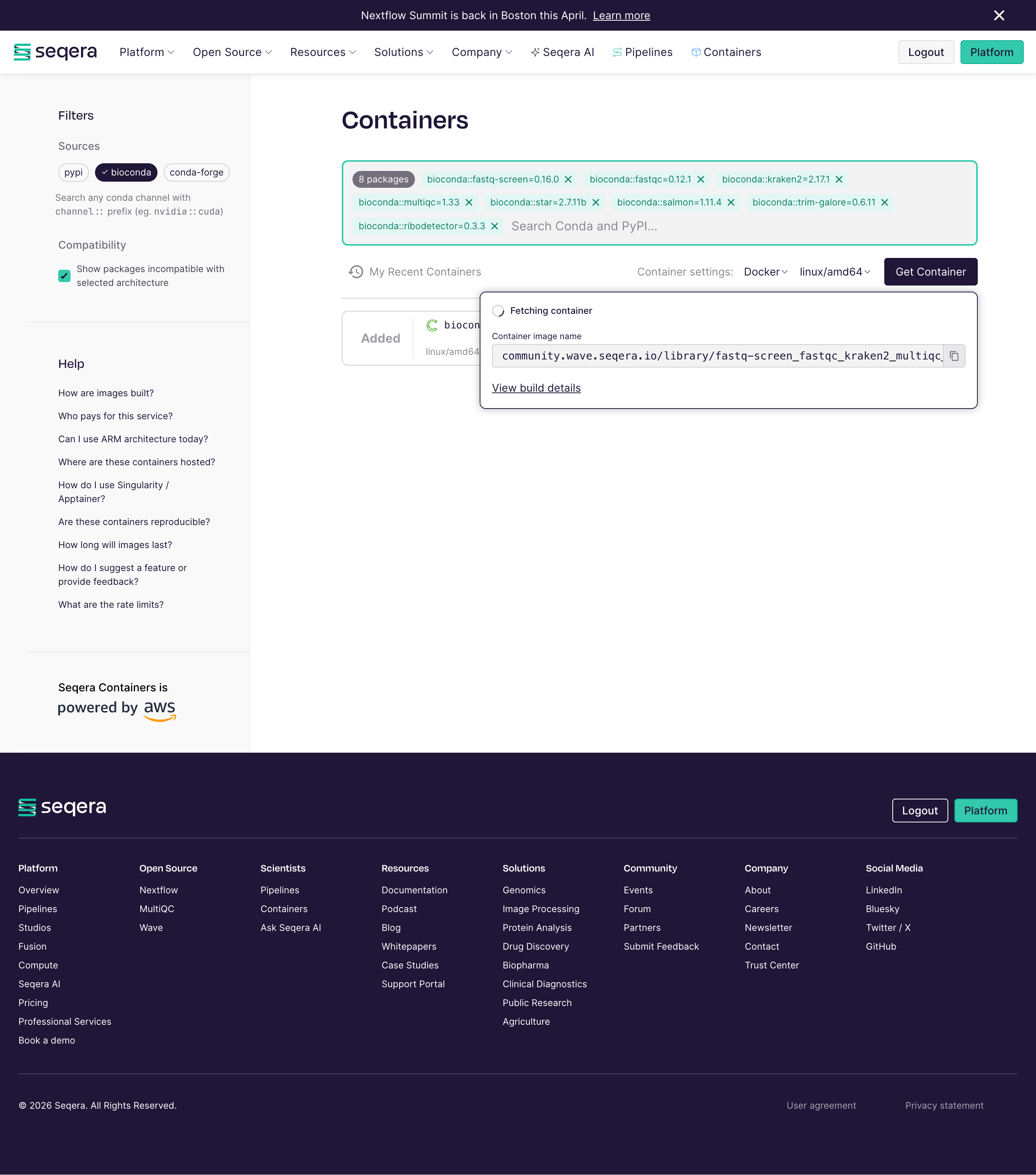
Seqera containers¶
Seqera Containers is a freely available resource that simplifies the container generation, allowing researchers to make a container for any combination of Conda and PyPI packages with the click of a button. It was launched in spring 2024 during the Nextflow Summit in Boston, and it is powered by Amazon Web Services and built on Wave, Seqera’s open-source container provisioning service.
Here you can see the webpage and make the containers that you need!